Dott.ssa Chiara Bianchi, Dott.ssa Martina Vitali, Dott. Marco Micillo, Dott. Francesco Vitali
Università degli studi di Ferrara
Azienda Ospedaliero-Universitaria di Ferrara – Arcispedale Sant’Anna
Presentiamo il caso di un paziente di 26 anni, senza comorbidità, con analisi genetica positiva per una mutazione del gene SCN5A, che va incontro ad un episodio di marcata instabilità emodinamica con temporanea perdita di coscienza e di polso. Il quadro elettrocardiografico del paziente e l’assenza di una cardiopatia strutturale depongono per un disturbo “atipico” del sistema di conduzione cardiaco associato alla mutazione del gene SCN5A. In considerazione della storia clinica e della mutazione di cui il paziente è portatore, si è deciso di procedere all’impianto di un pacemaker bicamerale definitivo.
Un ragazzo di 26 anni accede in Pronto Soccorso per un fugace episodio sincopale post-minzionale avvenuto nelle prime ore del mattino. Poco dopo aver ripreso conoscenza ha un secondo svenimento più prolungato associato a tremori diffusi. Viene soccorso dal padre, un infermiere del 118 che, non riuscendo a sentire il polso del figlio, comincia le manovre di rianimazione cardiopolmonare. Dopo circa mezzo minuto si verifica una ripresa spontanea di coscienza associata a flushing del volto e respiro russante. In quel momento viene documentata dal padre una frequenza cardiaca ritmica di circa 33-34 bpm.
All’elettrocardiogramma registrato in Pronto Soccorso il paziente presenta un blocco atrioventricolare di primo grado (intervallo PR: 280 ms) ed un blocco di branca destra completo (QRS: 160 ms).
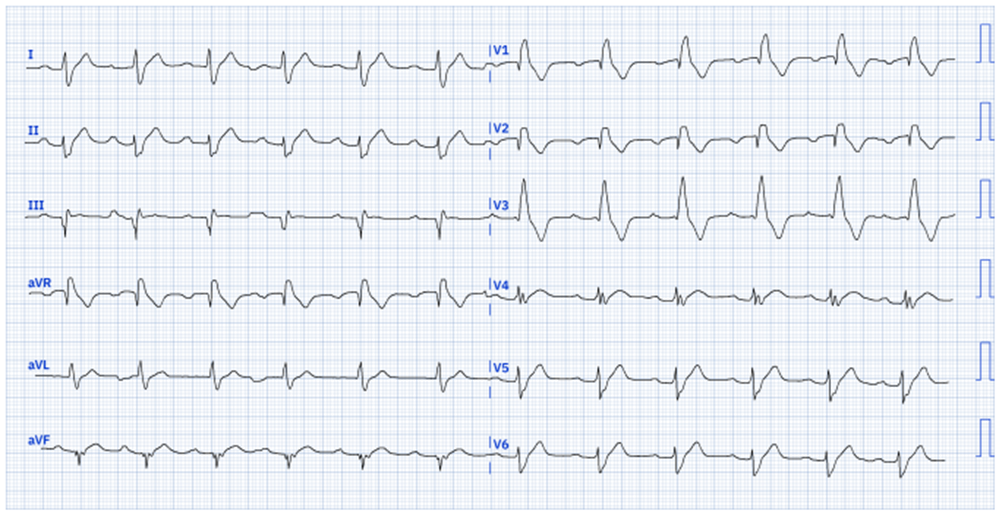
Raccogliamo l’anamnesi del ragazzo: è sempre stato bene, non ha particolari fattori di rischio cardiovascolare e fa molta attività sportiva. Gli è già capitato un’altra volta di perdere conoscenza per pochi secondi, circa due anni fa, durante un episodio di dolore addominale.
Il paziente è accompagnato da sua madre, una maratoneta professionista che ci racconta di avere impiantato un pacemaker da giovane a causa di un disturbo di conduzione per la quale è seguita insieme al figlio al nostro ambulatorio di Cardiogenetica. I due hanno eseguito un’analisi genetica un paio d’anni fa risultata positiva per una mutazione del gene SCN5A. Il figlio aveva eseguito anche un monitoraggio ECG-Holter delle 24 ore che testimoniava un ritmo sinusale con fasi notturne di bradicardia sinusale a 30 bpm circa e di blocco atrioventricolare di secondo grado tipo I. Aveva inoltre eseguito un ecocardiogramma transtoracico risultato normale ed una risonanza magnetica cardiaca che non evidenziava segni di cardiopatia strutturale, di metaplasia adiposa o di late gadolinium enhancement. Il suo intervallo QT si era sempre mantenuto nei limiti di norma ed il suo tracciato non aveva mai manifestato pattern di Brugada (nemmeno eseguendo l’ECG con il posizionamento degli elettrodi precordiali nel II, III e IV spazio intercostale).
Una volta raccolta questa anamnesi sospettiamo che il paziente abbia avuto una sincope cardiogena da blocco atrioventricolare parossistico. Ricoveriamo il ragazzo con monitoraggio telemetrico: durante l’osservazione non si verificano eventi aritmici ma solo occasionali tratti di bradicardia notturna con frequenza media di 40 bpm.
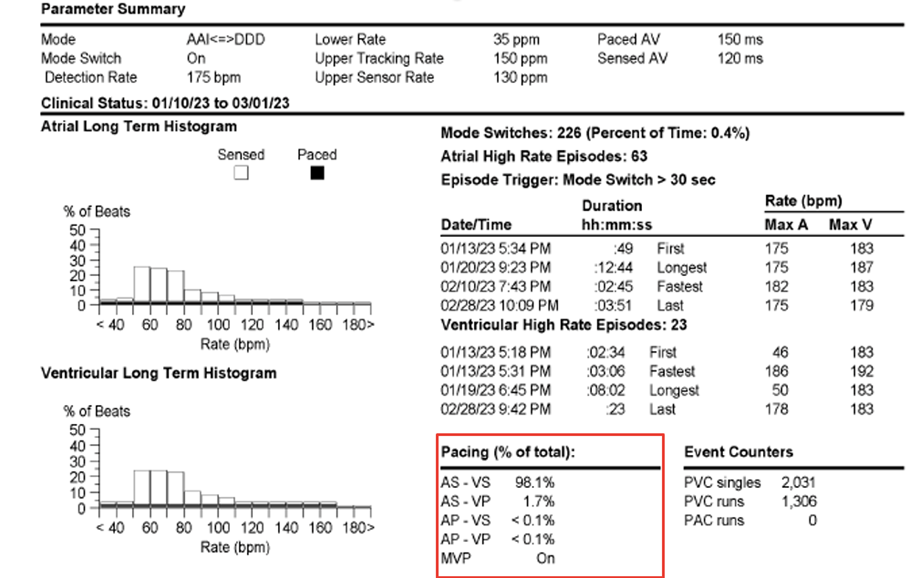
In considerazione della storia clinica e della mutazione di cui il paziente è portatore decidiamo di procedere all’impianto di un pacemaker bicamerale definitivo. Viene eseguita una stimolazione di branca e si decide di posizionare il dispositivo in sede sottomuscolare. Il dispositivo viene programmato in modalità AAI-DDD 35-150 bpm, con MVP attivo (per la natura attualmente parossistica dei blocchi), gestione automatica delle catture in entrambe le camere e monitoraggio remoto.
Attualmente il nostro paziente sta bene e ha ripreso la sua attività fisica regolare. Al controllo del device eseguito ad un mese dalla dimissione le percentuali di Atrial pacing sono < 0,1% e quelle di Ventricular pacing di circa 0.6%; i dati si confermano tali anche al monitoraggio remoto a quattro mesi dall’impianto. Tenendo conto del meccanismo patogenetico di malattia è verosimile che, nei prossimi anni, potremmo rilevare un aumento progressivo della percentuale di stimolazione del dispositivo.
Questo disturbo di conduzione geneticamente determinato (chiamato talvolta “disturbo progressivo familiare della conduzione cardiaca”, “disturbo di conduzione giovanile” o “sindrome di Lev-Lenègre”) è una patologia in cui la conduzione cardiaca viene progressivamente ostacolata nel tempo dalla fibrosi progressiva del sistema His-Purkinje. Ha un’età d’esordio variabile e si manifesta all’ECG con prolungamento progressivo dell’onda P, dell’intervallo PR e del segmento QRS, esitando talvolta in blocchi di branca destra o sinistra e/o blocchi atrioventricolari ingravescenti fino al blocco completo; può decorrere in modo asintomatico oppure manifestarsi con episodi di dispnea, vertigini, dolore addominale, oppure con sincopi a riposo o durante l’esercizio fisico, scompenso cardiaco o morte cardiaca improvvisa.
Questa sindrome è tipicamente, anche se non solo, associata a mutazioni loss of function del gene SCN5A. Tale gene si trova sul braccio corto del cromosoma 3 e codifica per la subunità α del canale del sodio voltaggio-dipendente che genera la corrente di depolarizzazione rapida (INa) responsabile della fase 0 del potenziale d’azione dei miocardiociti. La trasmissione è autosomica dominante a penetranza incompleta ed espressività variabile. Siamo soliti associare le mutazioni di questo gene alla Sindrome di Brugada, ma in realtà SCN5A presenta un’importante eterogeneità nelle sue mutazioni, che possono cambiare in diversi modi l’espressione del canale e le sue proprietà biofisiche, attraverso una loss of function oppure un gain of function. Ciò si traduce in molteplici possibili fenotipi clinici correlati alle sue mutazioni: la Sindrome di Brugada, la Sindrome del QT lungo sottoforma LQT-3, i disturbi di conduzione giovanili, la fibrillazione atriale idiopatica, la malattia del nodo del seno, l’arresto sinusale e la cardiomiopatia dilatativa. È inoltre possibile apprezzare quadri clinici di sovrapposizione (tipici delle mutazioni che provocano anche la Sindrome di Brugada) o di carattere aspecifico.
BIBLIOGRAFIA
1. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, et al. (2013) A Heterozygous Deletion Mutation in the Cardiac Sodium Channel Gene SCN5A with Loss- and Gain-of-Function Characteristics Manifests as Isolated Conduction Disease, without Signs of Brugada or Long QT Syndrome. PLoS ONE 8(6): e67963. doi:10.1371/journal.pone.0067963
2. Wilde AAM, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol. 2018 May;4(5):569-579. doi: 10.1016/j.jacep.2018.03.006. Epub 2018 May 2. PMID: 29798782.