Autori: Zefferino Palamà1,2, Martina Nesti3, Antonio Gianluca Robles2,4, Antonio Scarà5, Silvio Romano2, Elena Cavarretta6,7, Maria Penco2, Pietro Delise8, Mariano Rillo1, Leonardo Calò5 and Luigi Sciarra2,5
Commento a cura di: Lorenzo-Lupo Dei e Simona Minardi
1Elettrofisiologia, Casa di Cura “Villa Verde”, Via Golfo di Taranto, 22, Taranto, Italia
2 Dipartimento di Medicina Clinica, Sanità Pubblica, Scienze della vita e dell’ambiente, Università di L’Aquila, L’Aquila, Italia
3 Dipartimento Cardiovascolare e Neurologico, Ospedale San Donato, via Nenni, 20/22, Arezzo, Italia
4 Cardiologia, Ospedale “Di Venere”, Bari, Italia
5 Cardiologia, Policlinico Casilino, via Casilina, Roma 1049, Italia
6 Dipartimento di Scienze medico-chirurgiche e Biotecnologie , Università Sapienza , Latina, Italia
7 Cardiocentro Mediterranea, Napoli, Italia
8 Cardiologia, Ospedale P. Pederzoli, Peschiera Del Garda (VR), Italia
La fibrillazione atriale (FA) è la più comune tra le aritmie sostenute, colpisce circa il 3% della popolazione adulta ed è associata ad un aumentato rischio di ictus, scompenso cardiaco, riduzione della sopravvivenza e della qualità della vita.
Le tecniche di ablazione sono ad oggi considerate superiori alla terapia farmacologica nella strategia di controllo del ritmo (studi RAAFT, CABANA, EAST-AFNET4). Tuttavia, nonostante i progressi tecnologici e l’esperienza sempre maggiore degli operatori, i risultati post ablazione di FA sembrano aver raggiunto un plateau. Inoltre, sebbene la frequenza di complicanze sia bassa, è comunque rimasta invariata nel tempo. Per di più alcune complicanze, anche se rare, possono essere inaccettabili, come recentemente dimostrato nell’ESS–PRAFA snapshot.
Nell’ultimo ventennio sono stati messi in atto numerosi sforzi sul piano tecnologico e organizzativo per migliorare la qualità e la performance dei laboratori di elettrofisiologia ma pochi per personalizzare l’approccio al singolo paziente, che spesso intraprende un percorso standardizzato verso l’ablazione.
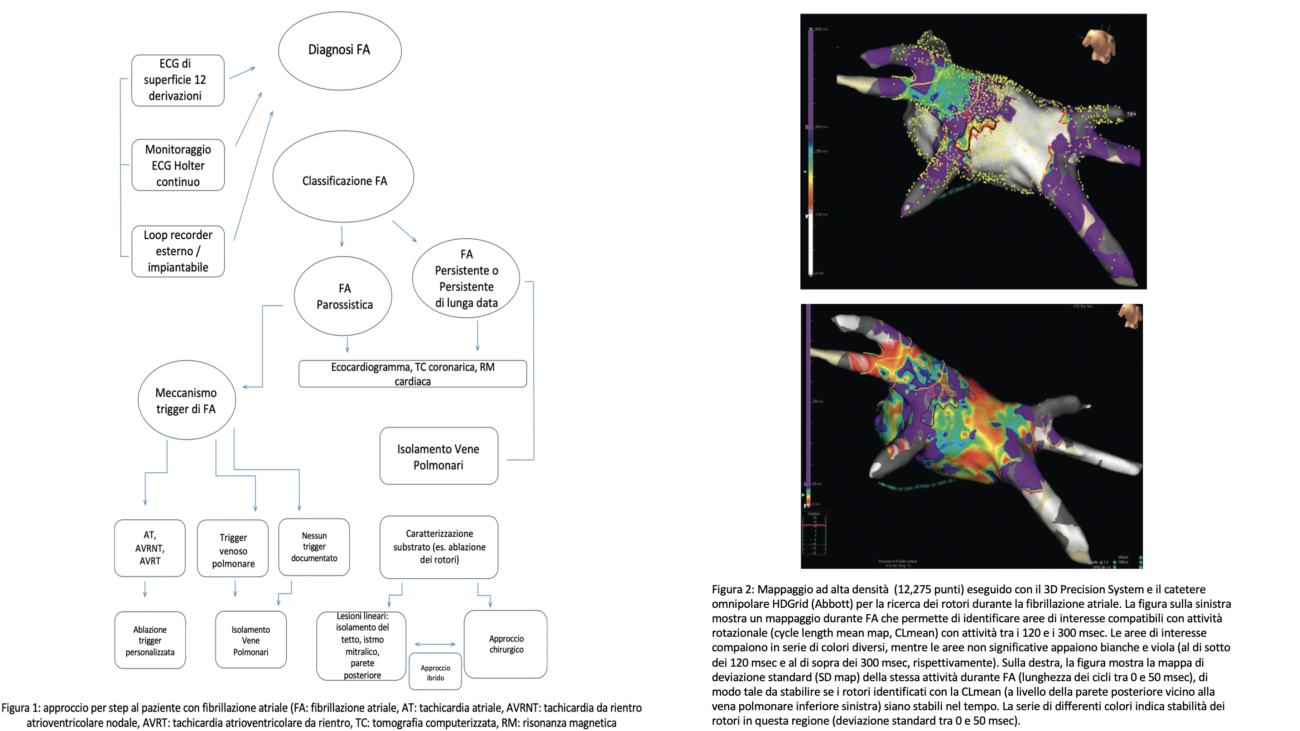
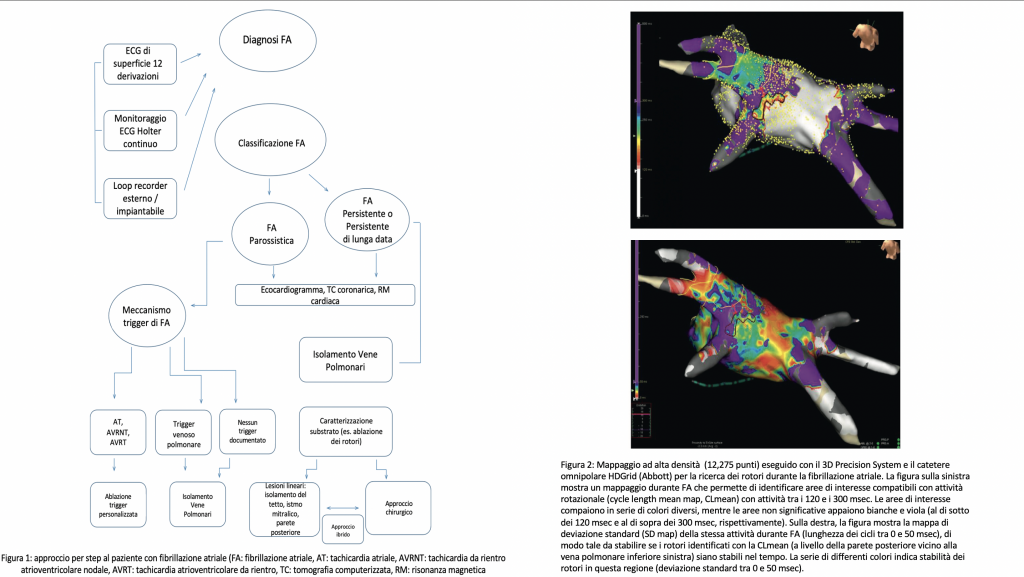
Al fine di migliorare il rapporto efficacia/rischio, il nostro gruppo propone una programmazione personalizzata della procedura di ablazione (fig.1), che selezioni, caso per caso, strategie di intervento differenti sulla base degli specifici meccanismi fisiopatologici dell’aritmia.
L’FA può essere classificata sulla base della durata dell’episodio in: parossistica (<7 giorni), persistente (>7 giorni, o che necessiti di una cardioversione a prescindere dalla durata) e permanente (ininterrotta, strategia di controllo del ritmo non prevista).
La fisiopatologia aritmica è descritta dal triangolo di Coumel: trigger, substrato aritmogeno e fattori modulanti.
Più frequentemente la forma di FA parossistica è trigger-relata mentre la persistente è sostenuta da uno specifico substrato. Per questo, nel setting di FA persistente è fondamentale la corretta caratterizzazione del substrato e dei meccanismi che sostengono l’aritmia al fine di programmare una strategia ablativa che vada oltre le vene polmonari. La forma parossistica è invece fortemente influenzata dalle comorbidità (ipertensione, diabete, obesità). In entrambi i casi, l’ablazione dell’aritmia non può prescindere da un preciso inquadramento clinico.
C’è tuttavia da tener conto del fatto che la correlazione tra classificazione e fisiopatologia è scarsa, senza dimenticare il ruolo del terzo attore del triangolo di Coumel, i fattori modulanti. Ad esempio, nei pazienti con FA vago-mediata esiste la possibilità di modificare l’influenza del sistema nervoso autonomo andando ad ablare i plessi gangliari intramurali, in aggiunta al trattamento ablativo standard.
Di volta in volta, è importante prendere in considerazione tutti gli strumenti a disposizione, che possono aiutare ad identificare i meccanismi che triggerano o mantengono l’FA. Quando non otteniamo abbastanza informazioni cliniche dai test non invasivi, potrebbe essere indicato, prima dell’ablazione, uno studio elettrofisiologico mirato ad indagare trigger diversi dalle vene polmonari, con la duplice finalità di raggiungere un migliore successo a lungo termine e di eseguire una procedura tecnicamente più semplice, con tassi di complicanze minori. Oggi l’evoluzione tecnologica permette di osservare il substrato con nuovi occhi: nuovi cateteri multipolari e tecniche di mappaggio 3D ad alta densità. Possono quindi essere proposte ablazioni più puramente anatomiche, con procedure chirurgiche, o meglio ibride, che facilitano il trattamento efficace di strutture difficili da raggiungere nel contesto di procedure completamente endocardiche (parete posteriore dell’atrio sinistro, legamento di Marshall, fascio di Bachmann).
L’approccio personalizzato, tuttavia, può risultare time-consuming e talvolta ritardate la strategia ablativa.
Ciò nonostante, la programmazione per step delle procedure di ablazione, adattata al paziente e alle caratteristiche della sua aritmia, dovrebbe essere routinaria in ogni laboratorio di elettrofisiologia.
L’accurata ricerca dei trigger nella FA parossistica ed il corretto riconoscimento del legame tra una possibile patologia cardiaca ed il substrato nella FA persistente potrebbe permettere di superare l’attuale plateau in termini di successo della procedura ablativa ed al contempo di minimizzarne le complicanze.
Andrea Faggiano 1, Marco Vicenzi 1, Stefano Carugo 1
1 Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico di Milano, Dipartimento di Medicina Interna, Unità Operativa di Cardiologia, Università di Milano, Italia.
Abstract
Presentiamo il caso clinico di una paziente di 70 anni con anamnesi di Sclerosi Sistemica a coinvolgimento multiorgano giunta alla nostra attenzione per effettuare un test da sforzo cardiopolmonare (CPET) per dispnea ingravescente. Il CPET mostrava una severa riduzione della capacità aerobica secondaria a spiccata limitazione cardiogena. L’ecocardiogramma evidenziava una severa ipertrofia ventricolare sinistra, asimmetrica, a prevalenza settale. Dall’anamnesi emergeva la familiarità per morte improvvisa, sindrome del tunnel carpale bilaterale, rottura non traumatica del capo-lungo del bicipite configurando così la difficile diagnosi differenziale fra tre entità cliniche: cardiopatia sclerodermica a fenotipo ipertrofico, sclerosi sistemica associata ad amiloidosi cardiaca, sclerosi sistemica associata a cardiomiopatia ipertrofica. L’approccio multimodale di imaging sequenziale, combinato all’esecuzione dell’analisi genetica ci permetteva di raggiungere la diagnosi di cardiomiopatia ipertrofica e di condurre lo screening genetico e fenotipico nei famigliari di primo grado della paziente.
CasoClinico
La paziente del nostro caso clinico è una donna di 70 anni affetta dal 1988 di Sclerosi Sistemica (SS) con coinvolgimento multiorgano (fenomeno di Raynaud, esofagopatia, coinvolgimento intestinale, artrite, calcinosi, teleangectasie). In anamnesi mai segnalato coinvolgimento sclerodermico cardiaco e/o ipertensione polmonare. Giungeva alla nostra attenzione tramite il centro unico di prenotazione regionale per l’esecuzione di un test ergometrico cardiopolmonare (CPET), richiesto dal centro di riferimento in considerazione della dispnea ingravescente per sforzi lievi (classe NYHA II-III) lamentata dalla paziente negli ultimi mesi.
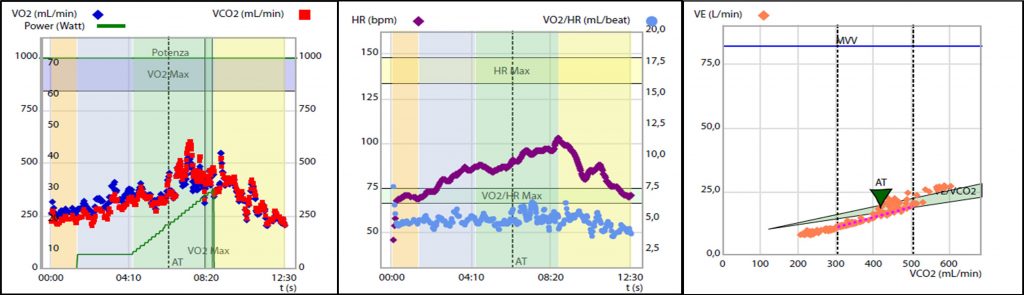
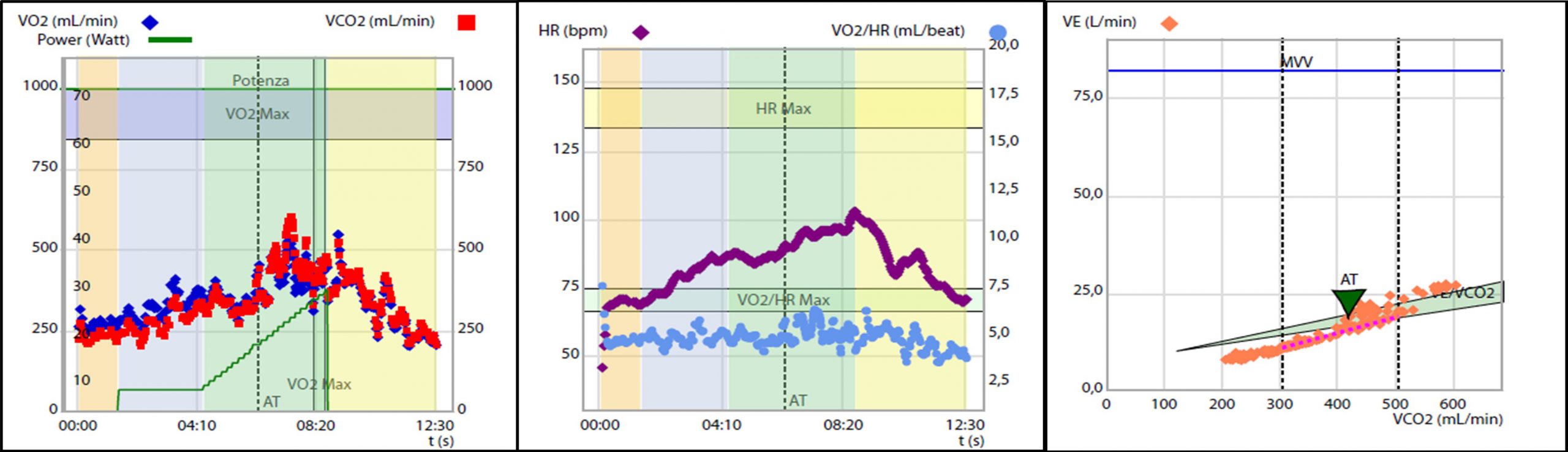
Figura 1. Test da sforzo cardiopolmonare eseguito durante la valutazione: 1A) Severa riduzione della capacità di esercizio aerobico certificata dal ridotto consumo di ossigeno al picco, pari a 9.9 ml/kg/min, ossia il 42% del predetto. 2A) Evidenza di severa limitazione cardiogena allo sforzo. Polso dell’ossigeno ridotto con plateau precoce e sostenuto, pari a 4 ml/battito, ossia il 60% del predetto. 3A) Inefficienza ventilatoria durante lo forzo, con slope del rapporto ventilazione -volume di anidride carbonica, VE/VCO2 slope pari a 39.9 ossia il 139% del predetto
Portava in visione l’ultimo ecocardiogramma, eseguito nel 2018, il quale mostrava lieve ipertrofia settale, normali dimensioni biventricolari, normale cinesi segmentaria e funzione sistolica globale, dilatazione atriale sinistra lieve, disfunzione diastolica di I° grado ed un basso rischio ecocardiografico di ipertensione polmonare. Le prove di funzionalità respiratorie recenti (2021) risultavano nella norma. L’elettrocardiogramma (ECG) basale effettuato prima del CPET rilevava segni di ipertrofia ventricolare sinistra con T negative in V3-V4-V5. Essendo la paziente di esile corporatura e tendente alla sarcopenia, il CPET veniva effettuato con una rampa blanda di incremento pari a 5 W/min.
L’esame veniva interrotto precocemente a 27 W per dispnea (BORG scale semplificata = 9) ed esaurimento muscolare (BORG scale semplificata = 8), risultava appena massimale per quoziente respirato (RQ = 1.1) e sottomassimale per frequenza cardiaca (FC = 103 bpm, 70% della massima frequenza cardiaca predetta). Dal CPET emergeva un quadro di severa riduzione della capacità di esercizio aerobico (consumo di ossigeno al picco, VO2 peak pari a 9.9 ml/kg/min, ossia il 42% del predetto. Figura 1 A) secondario a spiccata limitazione cardiogena (polso dell’ossigeno = 4 ml/battito, ossia il60% del predetto, con plateau precoce e sostenuto. Figura 1 B) associata ad inefficienza ventilatoria (slope del rapporto ventilazione – volume di anidride carbonica, VE/VCO2 slope = 39.9, ossia il 139% del predetto. Figura 1 C) in assenza di limitazione respiratoria.
In considerazione del quadro elettrocardiografico basale e del CPET si decideva di eseguire durante la stessa seduta un esame ecocardiografico (Panel 2) che mostrava una marcata ipertrofia asimmetrica del ventricolo sinistro, maggiore a carico del setto interventricolare (18 mm), in assenza di “systolic anterior movement” e di ostruzione dinamica all’efflusso, né in basale, né durante manovra di Valsalva ed in presenza di iniziale incremento del gradiente atrioventricolare destro (Tricuspid regurgitant Jet = 3 m/s).
Dati i reperti di imaging veniva ripercorsa l’anamnesi da cui emergeva la familiarità per morte improvvisa (sorella del padre morta improvvisamente a 58 anni) che poneva il forte sospetto di cardiomiopatia ipertrofica (HCM) sottostante. Inoltre, la paziente presentava in anamnesi l’intervento di tunnel carpale bilaterale e la rottura del capo-lungo del bicipite per traumatismo minore, entrambe red-flags per amiloidosi cardiaca in presenza di ipertrofia ventricolare sinistra (1).
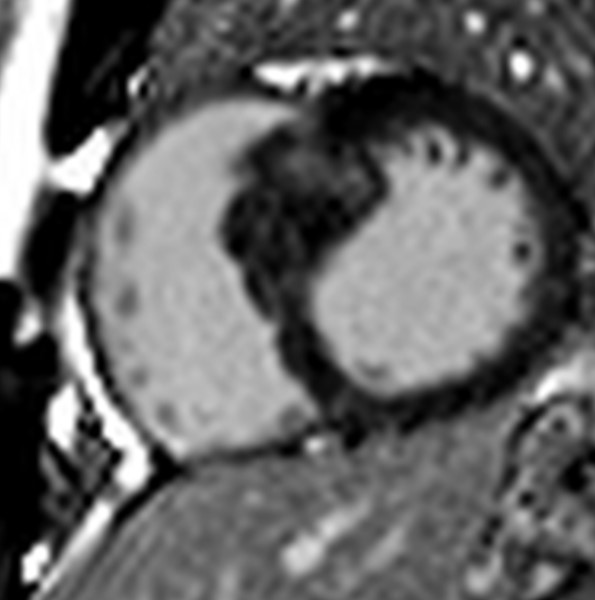
Figura 2. Ecocardiogramma transtoracico eseguito durante la valutazione che mostrava una severa ipertrofia ventricolare sinistra asimmetrica.
Inoltre, in considerazione della storia di SS con coinvolgimento multiorgano, una terza opzione possibile in grado di spiegare il quadro clinico della paziente si configurava nella cardiopatia sclerodermica a fenotipo ipertrofico.
Per escludere l’opzione clinicamente più urgente, ossia l’amiloidosi cardiaca, venivano effettuate la scintigrafia miocardica con tracciante osseo e l’elettroforesi con immunofissazione sierica ed urinaria. Questi esami risultavano negativi, permettendoci così di escludere l’opzione dell’amiloidosi cardiaca. L’esame diagnostico successivo scelto fu la risonanza magnetica cardiovascolare, la quale, nonostante fosse limitata in termini di qualità dalla claustrofobia della paziente, confermava la marcata ipertrofia del ventricolo sinistro con prevalente coinvolgimento del setto interventricolare e repertava multiple aree di late gadolinium enhancement a distribuzione “patchy”, in assenza di edema (Figura 3).
Tali reperti di imaging, nonostante tipici della HCM (2), sono anche aspetto peculiare del coinvolgimento cardiaco da accumulo fibrotico della SS (3). Pertanto, per facilitare la diagnosi differenziale fra cardiopatia sclerodermica e coesistenza di HCM + SS, dopo aver ricostruito tre generazioni di albero genealogico senza individuare eventi chiave oltre a quello sopra-citato, la paziente veniva sottoposta ad un test genetico con panel customizzato per i geni responsabili delle cardiomiopatie strutturali.
Figura 3. La risonanza magnetica mostra aree “patchy” intramiocardiche di late gadolinium enhancement con risparmio subendocardico, reperti compatibili sia con la cardiomiopatia ipertrofica che con la cardiopatia sclerodermica a fenotipo ipertrofico.
In attesa dell’esito della genetica, veniva effettuata la stratificazione aritmica mediante l’esecuzione di un holter ECG delle 48 risultato negativo per eventi aritmici ventricolari ripetitivi. Sia lo Score multi-parametrico (HCM-SCD risk score) consigliato dalle Linee Guida Europee sull’HCM (4) che l’approccio “single major risk criteria” suggerito dalla Linee Guida Americane (5) rivelavano la non indicazione all’impianto di defibrillatore impiantabile in prevenzione primaria in caso di HCM. L’esito dell’ analisi genetica mostrava la presenza in eterozigosi della variante c.2167C> G nell’esone 20 del gene myosin heavy chain 7 (MYH7), mutazione responsabile di circa il 15-25% dei casi di HCM (6). Pertanto, l’esecuzione dell’analisi genetica ci ha permesso, non solo di effettuare una diagnosi complessa, ma anche di avviare lo screening fenotipico e genetico a cascata ai familiari di primo grado della paziente, screening tuttora in corso.
Discussione e Conclusioni
Nella pratica clinica ci si trova quotidianamente ad affrontare il dilemma fra singola malattia in grado di spiegare una presentazione clinica complessa e la coesistenza contemporanea di più malattie responsabili. Sono due gli approcci clinico-filosofici contrapposti. Il primo, inconsciamente ispirato al “Rasoio di Guglielmo di Occam” (7), il quale suggerisce che la spiegazione più semplice sia quella da preferire e che pertanto sia più probabile che una sola malattia spieghi in toto la clinica del paziente. Il secondo approccio invece, sulla scia della Legge di Murphy (8) (“se qualcosa può andare storto, lo farà”) suggerisce che una manifestazione clinica complessa sia più probabilmente da imputare alla coesistenza di più malattie sottostanti. Nel caso clinico della nostra paziente tre erano le diagnosi differenziali principali: cardiopatia sclerodermica a fenotipo ipertrofico, sclerosi sistemica associata ad amiloidosi cardiaca, sclerosi sistemica associata a cardiomiopatia ipertrofica. In tutti e tre i casi si trattava di entità cliniche relativamente rare. In Nord Italia la prevalenza di SS è di 1:400 soggetti, di cui circa il 30% mostrano un coinvolgimento cardiaco ( in primis l’ipertensione arteriosa polmonare), ossia 1: 1250 individui (9). Il fenotipo ipertrofico della cardiopatia sclerodermica è una condizione clinica solo riportata in letteratura, di cui non è nota la prevalenza e la cui peculiarità anatomo-patologica principale è l’accumulo a mosaico del materiale fibrotico nel miocardio (10) (11). Invece, l’associazione probabilistica fra SS e amiloidosi cardiaca è stimabile attorno ad 1:10’000’000 soggetti (1), mentre quella fra SS ed HCM attorno ad 1: 200 000 soggetti (5). Soltanto un approccio di imaging multimodale sequenziale combinato all’esecuzione dell’analisi genetica ci ha permesso di effettuare una diagnosi complessa e di iniziare lo screening genetico e fenotipico dei famigliari di primo grado. Pertanto, nel caso da noi riportato, la Legge di Murphy ha avuto la meglio su Rasoio di Occam.
Bibliografia
1. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16).
2. Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part I, MRI appearances. Vol. 189, American Journal of Roentgenology. 2007.
3. Hachulla AL, Launay D, Gaxotte V, De Groote P, Lamblin N, Devos P, et al. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68(12).
4. Zamorano JL, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Vol. 35, European Heart Journal. 2014.
5. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020.
6. Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, Mccormack L, et al. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15(12).
7. Wildner M. In memory of William of Occam [16]. Vol. 354, Lancet. 1999.
8. Scott JP, Ayano C, Sulman CG, Ruiz JP. Murphy’s law and Murphy eyes. Vol. 32, Journal of Anesthesia. 2018.
9. Lo Monaco A, Bruschi M, La Corte R, Volpinari S, Trotta F. Epidemiology of systemic sclerosis in a district of northern Italy. Clin Exp Rheumatol. 2011;29(2 SUPPL. 65).
10. Moyssakis I, Papadopoulos DP, Anastasiadis G, Vlachoyannopoulos P. Hypertrophic cardiomyopathy in systemic sclerosis. A report of two cases. Clin Rheumatol. 2006;25(3).
11. Sogomonian R, Alkhawam H, Lee S, Chang D, Moradoghli Haftevani E. ID: 6: HYPERTROPHIC OBSTRUCTIVE CARDIOMYOPATHY IN THE SETTING OF SYSTEMIC SCLERODERMA. J Investig Med. 2016;64(4).
Le aritmie ventricolari rappresentano una criticità per la valutazione dell’idoneità sportiva negli atleti. L’iter diagnostico ideale di atleti agonisti con aritmie ventricolari complesse non è stato chiaramente definito. Lo scopo di questo studio è stato valutare le implicazioni cliniche della valutazione elettrofisiologica invasiva e della biopsia endomiocardica in atleti con aritmie ventricolari.
Sono stati valutati 227 atleti giunti a valutazione dopo essere stati squalificati a causa di aritmie ventricolari.
In questo studio, un work-up invasivo completo ha fornito elementi diagnostici aggiuntivi e potrebbe migliorare la valutazione dell’idoneità sportiva negli atleti che si presentano con aritmie ventricolari. Un’estesa valutazione invasiva potrebbe essere particolarmente utile quando i test non invasivi mostrano risultati poco chiari.
In questo studio di Dello Russo et al (1) è stata condotta un’analisi retrospettiva di atleti arruolati in due centri, all’Unità Operativa di Aritmologia, Centro di Cardiologia Monzino, IRCCS, Milano, Italia, e presso la Clinica di Cardiologia e Aritmologia, Azienda Ospedaliera-Universitaria “Ospedali Riuniti”, Ancona, Italia.
Da Febbraio 2010 a Settembre 2019, sono stati arruolati 227 atleti squalificati dalla partecipazione a gare sportive a causa di tachicardie ventricolari non sostenute (NSVTs), tachicardie ventricolari sostenute (SVT), complessi ventricolari prematuri (PVC) frequenti o PVC da sforzo di qualsiasi morfologia rilevate al monitoraggio ECG o al test ergometrico, o fibrillazione ventricolare (VF) rianimata.
L’iniziale valutazione diagnostica non invasiva comprendeva test ergometrico, ecocardiogramma, risonanza magnetica cardiaca (cMRI) e angio-TC coronarica (se sospetto di malattia coronarica aterosclerotica). Dopo gli esami non invasivi sono stati eseguiti, seguendo un protocollo istituzionale prestabilito, lo studio elettrofisiologico (EPS) ed il mappaggio elettroanatomico tridimensionale (EAM) in caso di diagnosi dubbia dopo gli esami non invasivi o in caso di diagnosi di certezza in previsione di ablazione trans-catetere.
La biopsia endomiocardica (EMB) guidata dall’EAM o dall’imaging in cMRI è stata invece eseguita in caso di incertezza diagnostica dopo i test non invasivi, EAM ed EPS. Inoltre, quando si sospettava clinicamente una miocardite, sono stati eseguiti EAM, EPS ed EMB per confermare la diagnosi. In caso di diagnosi clinica di cardiomiopatia aritmogena del ventricolo destro (ARVC), il work-up invasivo completo è stato eseguito ai fini di diagnosi differenziale tra ARVC e il cuore d’atleta.
L’età media della coorte era di 26 anni, e 44 soggetti (19%) erano di sesso femminile. La maggior parte (176 [78%]) erano atleti agonisti. PVC frequenti o PVC da sforzo erano le aritmie ventricolari più comuni alla presentazione (180 [79%]). Circa la metà degli atleti erano sintomatici (111 [49%]), più comunemente per cardiopalmo (82 [36%]), mentre 17 soggetti (8%) avevano una storia di sincope. Una storia familiare di morte cardiaca improvvisa (9 [4%]) o cardiomiopatia (7 [3%]) era presente in una minoranza di atleti.
Complessivamente, al termine della valutazione non invasiva, 81 pazienti avevano risultati normali, in 20 pazienti è stata identificata una miocardite clinicamente sospetta ed in 13 pazienti è stata diagnosticata clinicamente l’ARVC (9 con diagnosi borderline e 4 con diagnosi definitiva).
L’EAM è stato eseguito in 188 pazienti (83%). I risultati dell’EAM sono risultati anomali in 46 pazienti (24%), con evidenza di cicatrice miocardica (45) e/o potenziali tardivi (12).
L’EMB è stata eseguita in 42 atleti (15,2%) e ha permesso la formulazione di una diagnosi patologica in 32 atleti (76%). La diagnosi più comune era la miocardite (19 [45,2%]), mentre in 9 pazienti (23,8%) l’EMB ha permesso una diagnosi istopatologica di ARVC. È interessante notare che l’EMB ha consentito l’upgrade da diagnosi borderline a diagnosi definitiva di ARVC in 5 atleti.
Nel complesso, una cardiopatia strutturale è stata diagnosticata in 102 pazienti (45%) nell’intera popolazione di studio. Le patologie più comuni identificate erano miocardite (33 [15%]), cardiomiopatia dilatativa (23 [10%]) e ARVC (17 [8%]).
Tra i soggetti sottoposti a EMB, le diagnosi più frequenti erano miocardite (21 [50%]) e ARVC (13 [31%]). Quest’ultima interessava esclusivamente il ventricolo destro in 6 casi (46%), mentre 5 pazienti avevano interessamento biventricolare (39%) e 2 (15%) avevano un interessamento solo del ventricolo sinistro. Infine, 20 pazienti sono stati sottoposti ad impianto di ICD in prevenzione primaria (12) o secondaria (8).
La valutazione invasiva completa, inclusa l’EMB, ha consentito la riclassificazione diagnostica in 21 atleti (50%). La riclassificazione è stata particolarmente comune nel sottogruppo di pazienti con diagnosi non conclusiva dopo i test non invasivi (23 [87%]). La prevalenza di patologie cardiache nel campione è passato dal 30% (intervallo di confidenza (IC) al 95% 24-36%) quando si considerano solo i test non invasivi, al 37% (IC 95% 31-43%) dopo aver eseguito EAM ed EPS, ed infine al 45% (IC 95% 39-51%) dopo aver eseguito EMB.
DISCUSSIONE
Questi dati suggeriscono che: (1) dopo una valutazione non invasiva inclusa la cMRI, un certo grado
di incertezza diagnostica persiste in più di un terzo degli atleti con aritmie ventricolari, (2) un work-up diagnostico invasivo, inclusi EAM, EPS e EMB, potrebbe migliorare la definizione diagnostica, (3) in termini di riclassificazione, i test invasivi hanno avuto un impatto maggiore nei casi in cui i test non invasivi non hanno portato a una diagnosi definitiva, (4) questo lavoro ha fornito elementi importanti per la stratificazione del rischio aritmico e ha aiutato nella selezione dei pazienti candidati ad impianto di ICD.
Questi dati mostrano che non c’è stata riclassificazione diagnostica alla valutazione invasiva in atleti con risultati normali ai test non invasivi, confermando che i test non invasivi possono servire efficacemente come strumento per la stratificazione del rischio negli atleti e per selezionare i candidati ad ulteriori esami.
CONCLUSIONE
Un work-up diagnostico completo in questo studio ha fornito elementi diagnostici aggiuntivi e potrebbe migliorare la valutazione dell’idoneità sportiva negli atleti che si presentano con aritmie ventricolari. L’estesa valutazione con test invasivi potrebbe essere particolarmente utile quando i test non invasivi mostrano risultati poco chiari.
Dello Russo A, Compagnucci P, Casella M, Gasperetti A, Riva S, Dessanai MA, Pizzamiglio F, Catto V, Guerra F, Stronati G, Andreini D, Pontone G, Bonomi A, Rizzo S, Di Biase L, Capucci A, Natale A, Basso C, Fiorentini C, Zeppilli P, Tondo C. Ventricular arrhythmias in athletes: Role of a comprehensive diagnostic workup. Heart Rhythm. 2022 Jan;19(1):90-99.
Presentiamo il caso clinico di una paziente di 84 anni giunta alla nostra osservazione per STEMI laterale da restenosi di stent medicato su primo ramo marginale e con nota stenosi valvolare aortica severa. In prima e in seconda giornata successiva alla rivascolarizzazione percutanea (PCI), la paziente manifestava enterorragia massiva con conseguente anemizzazione. Il quadro veniva imputato alla presenza di angiodisplasia ileale. L’associazione tra stenosi aortica e sanguinamenti da angiodisplasia intestinale prende il nome di sindrome di Heyde, una condizione patologica per la quale ancora non esistono precise indicazioni per la gestione del quadro complessivo cardiologico e di diatesi emorragica.
CasoClinico
La paziente del nostro caso clinico è una donna di 84 anni affetta da ipertensione arteriosa, arteriopatia obliterante cronica periferica e insufficienza renale cronica al IV stadio. La paziente soffriva, inoltre, di fibrillazione atriale parossistica in terapia con anticoagulanti orali diretti (Apixaban) ed era stata sottoposta un anno prima a rivascolarizzazione percutanea (PCI) con impianto di stent medicato (DES) sul primo ramo marginale a seguito di un episodio di sindrome coronarica acuta NSTEMI. In anamnesi presentava inoltre storia di stenosi valvolare aortica severa di natura fibrocalcifica, per la quale la paziente aveva già iniziato il percorso di valutazione in previsione di sostituzione di valvola aortica transcatetere (TAVI).
A seguito della comparsa di dolore oppressivo precordiale scarsamente responsivo ai nitrati la paziente giungeva presso il nostro Pronto Soccorso. L’ elettrocardiogramma mostrava i segni della sindrome coronarica acuta STEMI laterale per cui veniva eseguita coronarografia in emergenza, con evidenza di malattia di un vaso coronarico da restenosi di DES precedentemente impiantato. L’occlusione veniva quindi trattata con angioplastica con palloncino (POBA) a seguito di carico di aspirina e di clopidogrel come da protocollo.
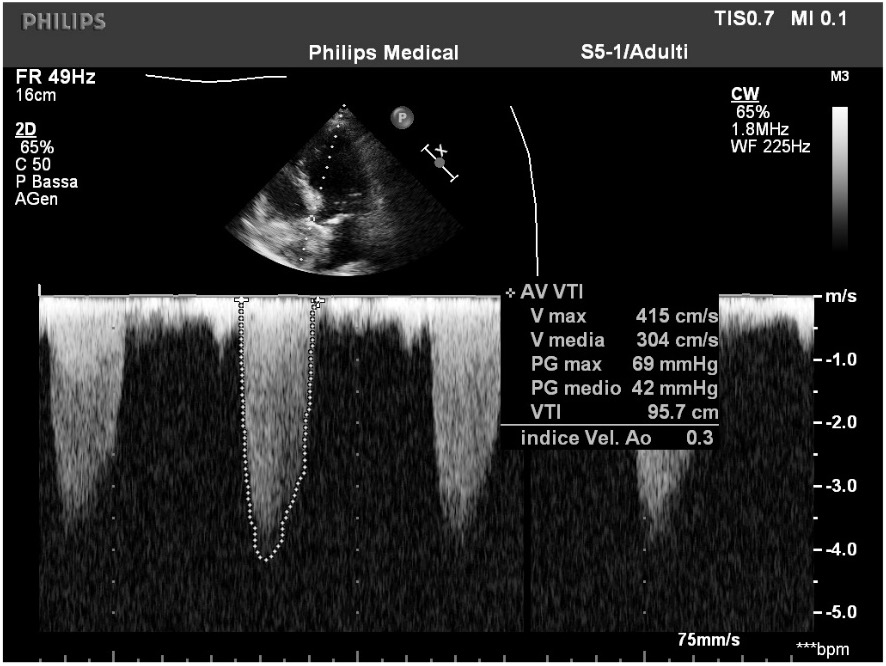
Figura 1 Ecocardiogramma eseguito all’ingresso: si confermano i valori emodinamici compatibili con stenosi aortica severa ad alto gradiente (Velocità di picco maggiore di 4 m/s, gradiente transvalvolare medio maggiore di 40 mmHg, FE conservata).
L’ecocardiogramma all’ingresso ( Figura 1) confermava la presenza di stenosi valvolare aortica severa di natura fibrocalcifica (Gradiente medio 42 mmHg, velocità di picco 4,15 m/s).
In prima e in seconda giornata successive alla PCI la paziente presentava episodi di enterorragia con conseguente grave anemizzazione, per cui eseguiva trasfusione di 4 sacche di emazie concentrate. Su indicazione del gastroenterologo venivano eseguite esofagogastroduodenoscopia (EGDS) e colonscopia, entrambe risultate negative per lesioni emorragiche, ma con evidenza di angiodisplasia ileale sanguinante.
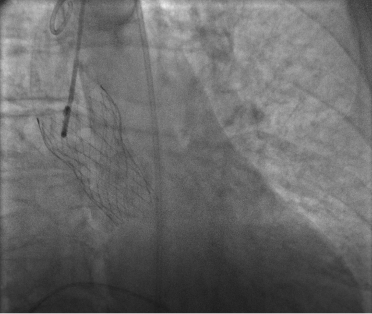
La concomitante presenza di stenosi valvolare aortica e angiodisplasie ileali è descritta in letteratura come Sindrome di Heyde. Nonostante la complicanza legata all’anemizzazione, a seguito del ripristino dei corretti valori emoglobinici, veniva posta indicazione all’esecuzione di TAVI (Figura 2). L’intervento non è stato gravato da complicanze acute. A seguito della procedura non si sono più verificati episodi di franche emorragie. Tuttavia, la paziente ha presentato un episodio di anemizzazione associata a febbre con la necessità di eseguire due emotrasfusioni, dopo le quali si è mantenuta su valori stabili di 10 mg/dL di emoglobina fino alla dimissione.
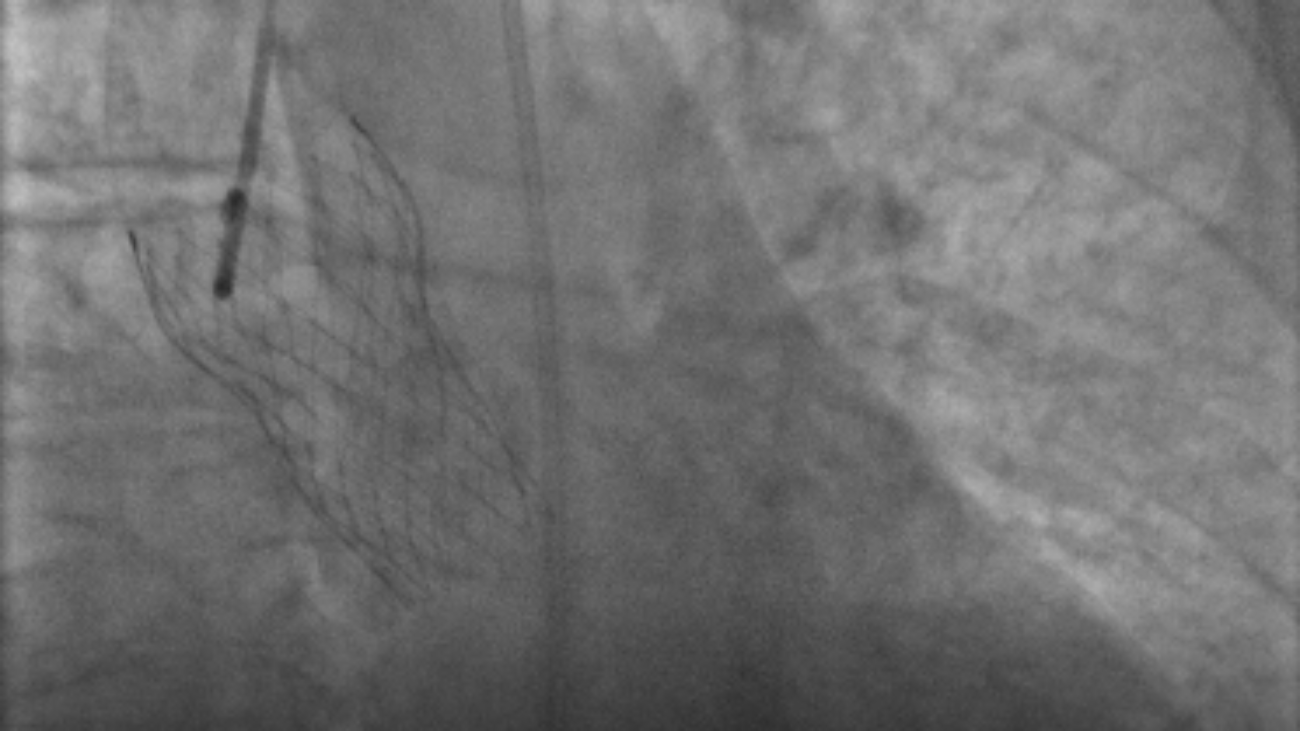
Figura 2: Immagine alla fluoroscopia di protesi valvolare aortica CoreValve.
La degenza della paziente è durata all’incirca un mese. Alla dimissione è stata prescritta unicamente terapia anticoagulante a base di Apixaban 5 mg/die in ragione dell’alto rischio emorragico.
Discussione
Si definisce sindrome di Heyde quella condizione patologica caratterizzata da associazione tra stenosi valvolare aortica fibrocalcifica e sanguinamenti gastrointestinali ricorrenti, descritta per la prima volta nel 1958.1 L’incidenza è maggiore in pazienti che presentano un’età in genere superiore ai 65 anni. Si stima che circa 1-3% di tutti i pazienti affetti da stenosi aortica moderata o severa manifestino sanguinamenti gastrointestinali clinicamente significativi.2 Uno studio condotto da Vincentelli et al. nel 2003 ha dimostrato che dal 20 al 70% dei pazienti con stenosi aortica presenta un deficit di multimeri ad alto peso molecolare del fattore di von Willebrand (VWF), per cui i dati sulla prevalenza della sindrome di Heyde sono probabilmente sottostimati.4 La patogenesi della sindrome è legata al flusso turbolento che si instaura a livello della valvola stenotica; la turbolenza favorisce infatti la degradazione del VWF tramite l’enzima ADAMTS 13. Il deficit di VWF, primario o secondario (noto, appunto, come malattia di Von Willebrand), è alla base di difetti dell’aggregazione piastrinica e degradazione del fattore VIII della coagulazione.3 È stato visto che i valori di VWF circolanti sono inversamente proporzionali al grado di severità della stenosi aortica. Inoltre, la morbilità e la mortalità dei pazienti con stenosi aortica e concomitante sindrome di Heyde sono peggiori rispetto a coloro che presentano la stenosi aortica isolata.5
Questi pazienti traggono beneficio dalla sostituzione della valvola difettosa? Dai dati in letteratura sembrerebbe che l’intervento di sostituzione della valvola aortica, eseguito sia attraverso via chirurgica (SAVR) che transcatetere (TAVI), riduca la probabilità di recidiva delle emorragie intestinali rispetto all’approccio conservativo6-9 Viene inoltre messa in luce la possibilità di valutare l’effetto della sostituzione valvolare tramite il dosaggio dei livelli di VWF.9
Conclusioni
La sindrome di Heyde è una condizione che colpisce dall’1 al 3% dei pazienti affetti da stenosi aortica moderato-severa e che attualmente non presenta da linee guida indicazioni specifiche per l’iter terapeutico. Gran parte degli studi che sono stati effettuati e i case report disponibili suggeriscono che la risoluzione della stenosi aortica comporta un miglioramento dell’interessamento gastrointestinale. Attualmente non esistono studi clinici randomizzati su larga scala capaci di fornire una chiara indicazione sul trattamento della stenosi valvolare aortica in quei pazienti con emorragie gastrointestinali ricorrenti e pertanto ci si affida all’esperienza e alla valutazione caso per caso. I pazienti affetti da stenosi aortica severa sono inoltre spesso pazienti fragili con comorbidità e maggior rischio di anemizzazione. In questi casi in cui coesiste elevato rischio trombotico a seguito di TAVI e rischio emorragico per le concomitanti angiodisplasie, grande attenzione deve essere posta nei riguardi della terapia antitrombotica/anticoagulante, con approfondita valutazione del rischio-beneficio.
2. Waldschmidt L, Drolz A, Heimburg P, et al. Heyde syndrome: prevalence and outcomes in patients undergoing transcatheter aortic valve implantation. Clinical research in cardiology : official journal of the German Cardiac Society 2021; 110(12): 1939-46.
3. Lourdusamy D, Mupparaju VK, Sharif NF, Ibebuogu UN. Aortic stenosis and Heyde’s syndrome: A comprehensive review. World journal of clinical cases 2021; 9(25): 7319-29.
4. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. The New England journal of medicine 2003; 349(4): 343-9.
5. Desai R, Parekh T, Singh S, et al. Alarming Increasing Trends in Hospitalizations and Mortality With Heyde’s Syndrome: A Nationwide Inpatient Perspective (2007 to 2014). The American journal of cardiology 2019; 123(7): 1149-55.
6. Dahiya DS, Kichloo A, Zain EA, Singh J, Wani F, Mehboob A. Heyde Syndrome: An Unusual Cause of Gastrointestinal Bleeding. Journal of investigative medicine high impact case reports 2021; 9: 2324709621997279.
7. Pyxaras SA, Santangelo S, Perkan A, et al. Reversal of angiodysplasia-derived anemia after transcatheter aortic valve implantation. Journal of cardiology cases 2012; 5(2): e128-e31.
8. King RM, Pluth JR, Giuliani ER. The association of unexplained gastrointestinal bleeding with calcific aortic stenosis. The Annals of thoracic surgery 1987; 44(5): 514-6.
9. Tsuchiya S, Matsumoto Y, Doman T, et al. Disappearance of Angiodysplasia Following Transcatheter Aortic Valve Implantation in a Patient with Heyde’s Syndrome: A Case Report and Review of the Literature. Journal of atherosclerosis and thrombosis 2020; 27(3): 271-7.
Enrico Guido Spinoni 1-2, Marco Mennuni 2, Andrea Rognoni 2, Leonardo Grisafi 1-2, Crizia Colombo 1-2, Veronica Lio 1-2, Giulia Renda 3, Melissa Foglietta 3, Ivan Petrilli 3, Damiano D’Ardes 3, Pier Paolo Sainaghi 1-2, Gianluca Aimaretti 1-2, Mattia Bellan 1-2, Luigi Castello 1-2, Gian Carlo Avanzi 1-2, Francesco Della Corte 1-2, Marco Krengli 1-2, Mario Pirisi 1-2, Mario Malerba 1-4, Andrea Capponi 2, Sabina Gallina 3, Sante Donato Pierdomenico 3, Francesco Cipollone 3, Giuseppe Patti 1-2, COVID-UPO Clinical Team.
Affiliations:
1 Università del Piemonte Orientale, Novara; 2 AOU Maggiore della Carità, Novara; 3 Università Gabriele d’Annunzio, Chieti-Pescara; 4 Ospedale Sant’Andrea, Vercelli.
Commentary
Atrial fibrillation (AF) shares with CoronaVirus Infective Disease (COVID-19) various prevalent cardiovascular (CV) co-morbidities (1-2). The occurrence of AF in patients with COVID-19 and its impact on outcome have not been specifically evaluated yet.
We investigated the incidence and prognostic impact of different subgroups of AF (historical and new onset) in consecutive patients hospitalized for COVID-19. A total of 637 patients, admitted in three major Italian hospitals, were included. In-hospital outcomes were investigated, choosing as primary endpoint the incidence of all-cause mortality and as secondary endpoints: the occurrence of CV death, non-CV death and severe acute respiratory distress syndrome (ARDS).
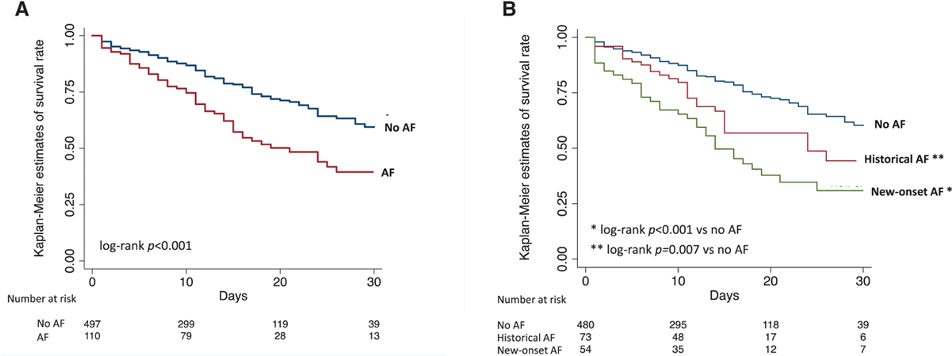
We included a total of 637 patients, 503 (79%) patients with stable sinus rhythm, and 134 (21%) in-hospital AF (historical in 79 patients and new-onset in 55). Patients with AF were older and presented a higher prevalence of various comorbidities: arterial hypertension, diabetes, cardiomyopathy, peripheral artery disease, chronic kidney disease, and chronic obstructive pulmonary disease. Our study outlined higher in-hospital mortality in patients with AF (44.4% versus 22.1% in those without, P=0.001); 30-day estimated survival rates by Kaplan-Meier method were 39.6% (95% CI, 27.8%–50.8%) versus 59.4% (51.4%–66.5%), respectively (log-rank P<0.001; Figure panel A). We performed logistic regression analysis, including demographic
factors, comorbidities, laboratory findings, and in-hospital treatments. At multivariate analysis the occurrence of AF was significantly associated with an increased risk of all-cause death (OR 2.44, 95% CI 1.18-5.07; p=0.016), CV death (OR 3.26, 95% CI 1.2–9.5; p=0.03) and severe ARDS (OR 1.96, CI 95% 1.07-3.6; p=0.03).
Patients with new-onset AF showed an increased incidence of in-hospital death (49.1% versus 36.7%), cardiovascular mortality (14.6% versus 5.1%), and ARDS (49.1% versus 29.7%)
compared with those with historical AF. The 30-day estimated survival rates were 44.3% (95% CI, 27.7%–59.6%) in patients with historical AF (log-rank p=0.007 versus no AF) and 30.8% (17.4%–45.2%) in those with new-onset AF (log-rank p<0.001 versus no AF; Figure [B]). Using patients without AF as reference, a stepwise increase in the risk of all-cause death across patients with historical AF (adjusted OR 1.26, 0.58-2.74) and those with new onset AF (adjusted OR 3.34, 1.54-7.25) was demonstrated.
Our study suggests the hypothesis that new-onset of AF in patients hospitalized for COVID-19 represent an independent predictor oin-hospital mortality, CV death, and more severe clinical pattern. In such setting, new-onset AF may represent potential clinical marker of adverse outcomes, as it is associated by higher degree of inflammatory and hypoxemic viral insult.
In conclusion, our study support the hypothesis that in patients hospitalized for COVID-19, the occurrence of AF is frequent and is independently associated with adverse outcome, including increased all-cause and cardiovascular mortality.
Figure: Kaplan-Meier curves at 30 days. Estimates of survival stratified by presence/absence of atrial fibrillation (AF; A) and by AF subtypes (B) are illustrated.
References:
1. Palmieri L, Vanacore N, Donfrancesco C, Lo Noce C, Canevelli M, Punzo O, Raparelli V, Pezzotti P, Riccardo F, Bella A, et al.; Italian National Institute of Health COVID-19 Mortality Group. Clinical characteristics of hospitalized individuals dying with COVID-19 by age group in Italy.J Gerontol A Biol Sci Med Sci. 2020; 75:1796–1800.
2. Inciardi RM, Adamo M, Lupi L, Cani DS, Di Pasquale M, Tomasoni D, Italia L, Zaccone G, Tedino C, Fabbricatore D, et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy.Eur Heart J. 2020; 41:1821–1829.
1 Cardiology Unit, Department of Medical and Surgical Specialties, Radiological Sciences and Public Health, University of Brescia, Brescia, Italy2 Cardiology Department, Faculty of Medicine, Alexandria University, Alexandria, Egypt
Abstract
L’ipertensione polmonare è caratterizzata da un aumento delle pressioni arteriose polmonari con dilatazione dell’arteria polmonare e del ventricolo destro. Riportiamo il caso di una donna di 47 anni affetta da ipertensione polmonare in terapia dal 2018 e ricoverata in cardiologia per peggioramento della dispnea.
La TC torace eseguita durante il ricovero ha mostrato un rapido aumento delle dimensioni dell’arteria polmonare (65 mm nel 2019, 76 mm nel 2021). Alla coronarografia è stata evidenziata una compressione ab estrinseco del tronco comune, trattata mediante stent.
La procedura è stata ben tollerata e nessun nuovo sintomo clinico è stato riferito a 3 mesi di follow-up.
Case report
L’ipertensione polmonare è una patologia caratterizzata dalla presenza di elevate pressioni arteriose polmonari (PAP), con dilatazione e disfunzione ventricolare destra.
Riportiamo il caso di una donna di 47 anni con diagnosi di ipertensione arteriosa polmonare primaria nota dal 2018, diagnosticata mediante cateterismo cardiaco destro (pressione arteriosa polmonare sistolica, PAPs 107 mmHg; PAP media, PAPm 69 mmHg; PAP diastolica, PAPd 50 mmHg). Nello stesso 2018 durante il test del cammino di 6 minuti (6MWT) la paziente aveva riferito dispnea dopo 4 minuti per una performance di 380 m.
Inizialmente, era stata impostata una terapia con Macitentan e Sildenafil e l’anno successivo (2019), è stato introdotto il Selexipag visto il peggioramento della dispnea e della performance al 6MWT (320 m con dispnea a 3 minuti).
Nel gennaio 2021 si è presentata in PS per peggioramento della dispnea, con successivo ricovero in Cardiologia.
L’elettrocardiogramma ha mostrato ritmo sinusale, senza alterazioni patologiche significative. L’ecocardiogramma transtoracico (TTE) ha, invece, evidenziato una importante dilatazione ed ipocinesia del ventricolo destro (diametro basale 60 mm, TAPSE 16 mm) con PAPs stimata di 90 mmHg, minimo rigurgito tricuspidalico e moderato rigurgito polmonare. La frazione di eiezione ventricolare sinistra è risultata nella norma (60%) con movimento paradosso del setto interventricolare.
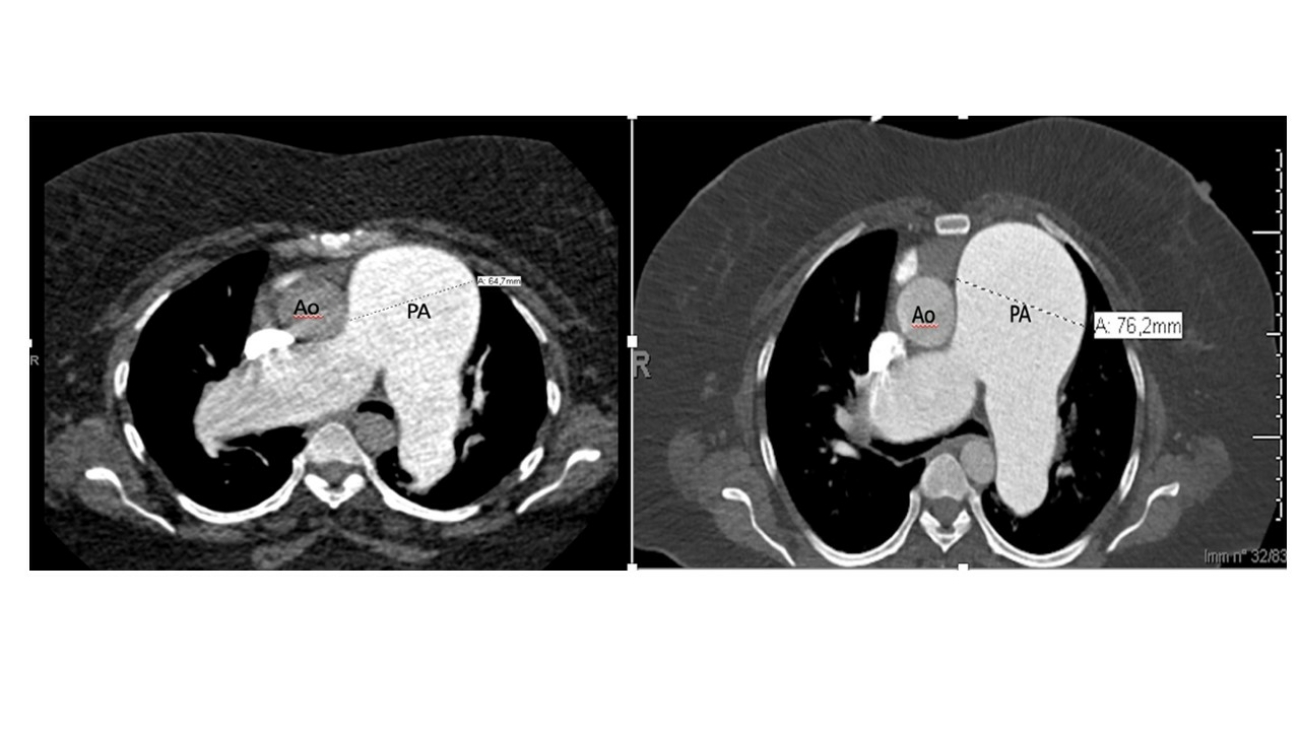
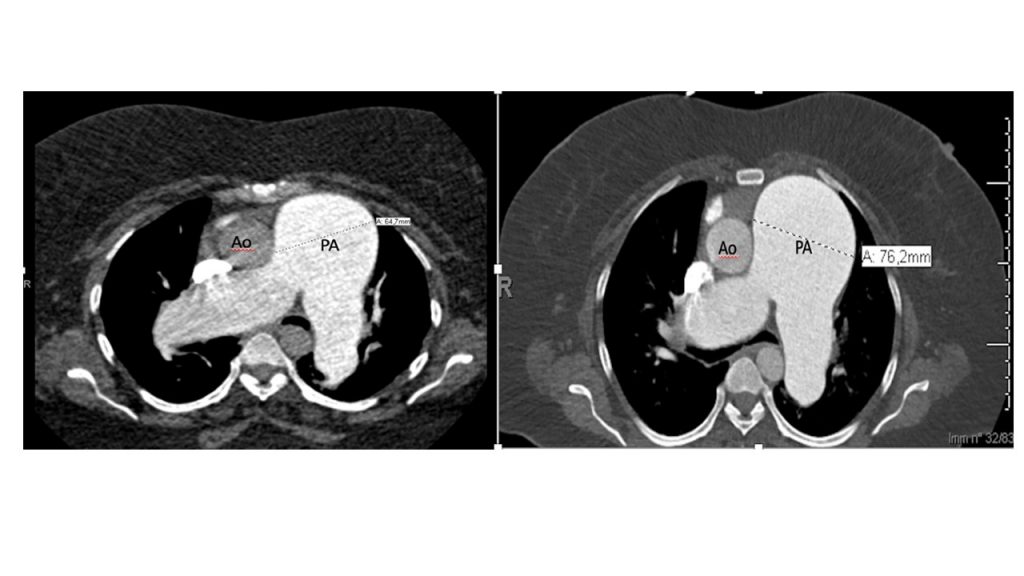
La TC eseguita ha escluso la presenza di embolia polmonare ma ha evidenziato una notevole dilatazione dell’arteria polmonare, in netto incremento rispetto al controllo precedente (65 mm nel 2019, 76 mm nel 2021), e dei suoi rami principali (50 mm contro 47 mm nel ramo destro, 42 mm contro 36 mm nel ramo sinistro) (Fig.1).
Fig 1: A sinistra: dimensioni TC dell’arteria polmonare nel 2019 (64,7 mm). A destra, dimensioni TC dell’arteria polmonare nel 2021 (76,2 mm).PA (Pulmonary artery), Ao (Aorta).
Durante il ricovero, è stato eseguito nuovamente il cateterismo cardiaco destro nel sospetto di peggioramento dell’ipertensione polmonare con evidenza di PAPs 120 mmHg e PAWP (pressione capillare polmonare) 100 mmHg.
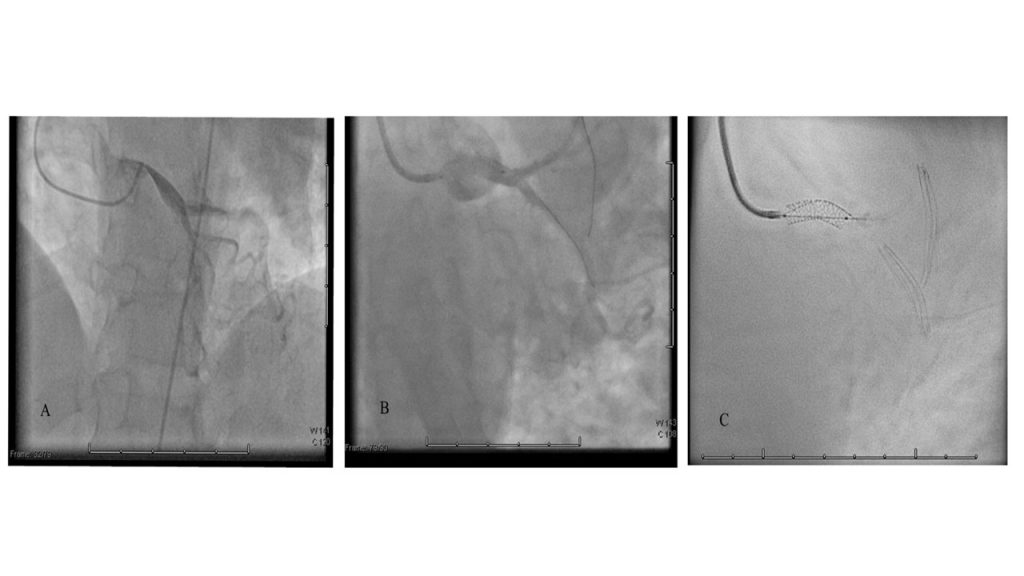
Nel sospetto di una compressione del tronco comune è stata posta indicazione ad angiografia coronarica, con effettivo riscontro di una compressione ab estrinseco del tronco comune dell’80% in assenza di placche ateromasiche. Il tronco comune è stato quindi valutato mediante ecografia intravascolare (IVUS), che ha confermato la presenza di un significativo restringimento luminale secondario alla compressione esterna. Considerando la significativa compressione esterna del tronco comune in una paziente con dispnea in peggioramento ed in assenza di dolore toracico o sincope, abbiamo deciso di eseguire un intervento coronarico percutaneo (PCI) con uno stent a rilascio di farmaco (DES) non protetto a livello del tronco comune con un buon risultato finale (Fig. 2). Non si sono verificate complicazioni durante e dopo la procedura e la paziente è stata dimessa dopo 48 ore in buone condizioni cliniche. Al follow-up a 3 mesi presso l’ambulatorio di ipertensione polmonare non ha riportato nessun nuovo sintomo.
Fig. 2: A. Coronarografia con evidenza stenosi del tronco comune dovuta a compressione estrinseca. B-C: Percutaneous coronary intervention (PCI) with unprotect drug-eluting stent (DES) su tronco comune
Discussione
La paziente ha presentato un rapido aumento delle dimensioni dell’arteria polmonare con compressione ab estrinseco del tronco comune senza sintomi se non un peggioramento della dispnea. Pertanto, il trattamento con PCI con DES sul tronco comune è stato mirato non solo a prevenire ulteriori sintomi ma anche la morte improvvisa, considerando soprattutto la rapida dilatazione dell’arteria polmonare (11 mm in 16 mesi) in una paziente con terapia già ottimizzata per l’ipertensione polmonare.
Questa è stata la prima volta in cui nel nostro centro è stata trattata una compressione ab estrinseco del tronco comune mediante PCI con DES nonostante l’assenza di angina o perdita di coscienza.
Secondo la letteratura, l’ipertensione arteriosa polmonare (PAH) è responsabile della compressione del tronco comune nel 5-19% dei pazienti, specialmente in quelli con PAH [1-2]. Il meccanismo è dovuto alla dilatazione della arteria polmonare. Tuttavia, la compressione del tronco comune dovuto alla dilazione dell’arteria polmonare è più spesso associata a cardiopatie congenite, in particolare ad un difetto del setto interatriale, del setto ventricolare, al dotto arterioso pervio o alla tetralogia di Fallot. L’ischemia miocardica significativa che consegue la compressione del tronco comune dipende sia dal grado di compressione del vaso stesso che dall’angolo che forma con il seno sinistro di Valsalva (soprattutto se inferiore a 30°). Anche il rapporto tra arteria polmonare e aorta superiore o uguale a 2 è considerato un fattore di rischio addizionale per la compressione del tronco comune [3-5]. Galiè et al [6] hanno dimostrato che un diametro dell’arteria polmonare di almeno 40 mm rappresenta il miglior predittore di stenosi del tronco comune del 50% o superiore.
Ad oggi, vi è una scarsità di protocolli per la gestione della compressione del tronco comune nell’ambito della PAH. Il bypass aortocoronarico e l’impianto di stent non protetto sono le uniche strategie attualmente utilizzate. Data l’elevata mortalità chirurgica nei pazienti con PAH, lo stent è, in genere, preferito come strategia di rivascolarizzazione di scelta e diversi autori hanno riportato risultati positivi in questo tipo di pazienti [7].
Conclusione
Nel caso descritto, dato il rapido aumento di dimensioni dell’arteria polmonare in paziente con nota ipertensione polmonare, la compressione ab estrinseco del tronco comune è stata trattata mediante stent per prevenire gli eventi avversi, in accordo con la letteratura.
Sarà necessario nei prossimi anni implementare un algoritmo di screening per valutare la progressione della dilatazione dell’arteria al fine di offrire un tempestivo trattamento per evitare la compressione del tronco comune, indipendentemente dai sintomi[8].
REFERENCES
Kothari SS, Chatterjee SS, Sharma S, Rajani M, Wasir HS. Left main coronary artery compression by dilated main pulmonary artery in atrial septal defect. Indian Heart J 46: 165-167, 1994.
Mesquita SM, Castro CR, Ikari NM, Oliveira SA, Lopes AA. Likelihood of left main coronary artery compression based on pulmonary trunk diameter in patients with pulmonary hypertension. Am J Med 116: 369-374, 2004.
Lee MS, Oyama J, Bhatia R, Kim YH, Park SJ. Left main coronary artery compression from pulmonary artery enlargement due to pulmonary hypertension: a contemporary review and argument for percutaneous revascularization. Catheter Cardiovasc Interv 76: 543-550, 2010.
Doyen D, Moceri P, Moschietto S, et al. Left main coronary artery compression associated with primary pulmonary hypertension. J Am Coll Cardiol. 2012; 60: 559.
Dodd JD, Maree A, Palacios I, de Moor MM, Mooyaart EA, Shapiro MD, Ferencik M, Brady T, Abbara S, Cury RC, Hoffmann U. Images in cardiovascular medicine. Left main coronary artery compression syndrome: evaluation with 64-slice cardiac multidetector computed tomography. Circulation. 2007 Jan 2;115(1):e7-8. doi: 10.1161/CIRCULATIONAHA.106.645622. PMID: 17200448.
Galiè N, Saia F, Palazzini M, Manes A, Russo V, Bacchi Reggiani ML, Dall’Ara G, Monti E, Dardi F, Albini A, Rinaldi A, Gotti E, Taglieri N, Marrozzini C, Lovato L, Zompatori M, Marzocchi A. Left Main Coronary Artery Compression in Patients With Pulmonary Arterial Hypertension and Angina. J Am Coll Cardiol. 2017 Jun 13;69(23):2808-2817. doi: 10.1016/j.jacc.2017.03.597. PMID: 28595696.Ogiso M, Serizawa N, Kamishima K, Yamaguchi J, Hagiwara N. Percutaneous coronary intervention for left main compression syndrome due to severe idiopathic pulmonary arterial hypertension: one year follow-up using intravascular imaging. Intern Med. 2015;54(7):801-4.
Fujiwara K, Naito Y, Higashiue S, Takagaki Y, Goto Y, Okamoto M, Yoshida S, Sekii H, Tomobuchi Y. Left main coronary trunk compression by dilated main pulmonary artery in atrial septal defect. Report of three cases. J Thorac Cardiovasc Surg. 1992; 104:449–452.
Labin JE, Saggar R, Yang EH, Lluri G, Sayah D, Channick R, Ardehali A, Aksoy O, Parikh RV. Left main coronary artery compression in pulmonary hypertension. Catheter Cardiovasc Interv. 2020 Nov 25. doi: 10.1002/ccd.29401. Epub ahead of print. PMID: 33241630.
Domenico Simone Castiello1, Domenico Angellotti1, Fiorenzo Simonetti1, Nicola Verde1, Lina Manzi1, Christian Basile1, Alfonsina Chirico1, Carlo Carbone1.
1Dipartimento di Scienze Biomediche Avanzate, Università Federico II di Napoli
Abstract
La degenerazione di bioprotesi valvolare è una delle complicanze più temute in seguito a sostituzione valvolare per via chirurgica o trans-catetere. In questo report presentiamo il caso clinico di una paziente di 80 anni con anamnesi di precedente intervento cardiochirurgico con impianto di bioprotesi aortica e mitralica con associata plastica tricuspidalica (2018) che, dopo circa 3 anni di buone condizioni di salute, presentava dispnea progressiva. La paziente accedeva quindi in PS dove, in seguito ad ecocardiogramma trans-esofageo, veniva posta diagnosi di insufficienza mitralica severa causata da degenerazione della bioprotesi mitralica precedentemente impiantata. Dato l’elevato rischio chirurgico si poneva indicazione ad impianto trans-catetere eterotopico di bioprotesi aortica in posizione mitralica per via trans-settale.
Caso clinico
Presentiamo il caso clinico di una paziente di 80 anni con i seguenti fattori di rischio cardiovascolare: ipertensione arteriosa, dislipidemia, pregressa abitudine tabagica ed affetta da ipotiroidismo e BPCO. In anamnesi la paziente è stata sottoposta precedentemente ad intervento cardiochirurgico (2018) durante il quale venivano eseguite contestualmente impianto di bioprotesi aortica (Saint Jude Medical Epic 21 mm) per evidenza di stenosi aortica severa, impianto di bioprotesi mitralica (Saint Jude Medical Epic 29 mm) per insufficienza mitralica severa ed associata plastica della valvola tricuspide (con tecnica De Vega) per insufficienza tricuspidale severa.
Da quel momento la paziente riferiva uno stato di buona salute fino al dicembre 2021 quando, per insorgenza di dispnea progressiva ed ingravescente, si recava in PS dove veniva sottoposta ad ecocardiogramma transtoracico, dal quale si evidenziava insufficienza mitralica severa. Si procedeva quindi ad ecocardiogramma transesofageo che mostrava degenerazione e fissurazione del lembo postero-laterale della bioprotesi mitralica.
In considerazione dell’elevato rischio chirurgico della paziente, in seguito a discussione del caso clinico in Heart Team, si poneva indicazione ad impianto eterotopico di bioprotesi aortica Edwards-Sapien 3 in posizione mitralica per via trans-settale. La paziente veniva quindi trasferita presso l’UTIC dell’AOU Federico II di Napoli per il prosieguo dell’iter diagnostico-terapeutico.
Al momento del ricovero la paziente si presentava in condizioni emodinamiche stabili, asintomatica per angor e palpitazioni, lievemente dispnoica a riposo. L’esame obiettivo cardiaco evidenziava soffio olosistolico III/VI Levine su tutti i focolai, principalmente sul focolaio mitralico; l’esame obiettivo toracico rivelava MV aspro diffusamente, con crepitii basali bilaterali; l’esame obiettivo generale evidenziava edemi declivi improntabili bilateralmente. L’elettrocardiogramma mostrava ritmo sinusale a FC di 65 bpm, con intervalli PR e QTc nei limiti. Gli esami ematochimici erano nella norma con conservata funzionalità renale.
Al ricovero l’ecocardiogramma transtoracico evidenziava malfunzione della bioprotesi in sede mitralica con lacerazione ed eversione del lembo postero-laterale determinante insufficienza mitralica severa (EROA: 0.6 cm2, volume rigurgitante: 95 mL). Il ventricolo sinistro di normali dimensioni cavitarie con conservata cinesi globale e segmentaria (FE: 58%). Bioprotesi in posizione aortica normo-funzionante. Esiti di plastica della valvola tricuspide con insufficienza residua di grado moderato da cui si stimava severo aumento della pressione arteriosa polmonare sistolica (PAPS: 80 mmHg).
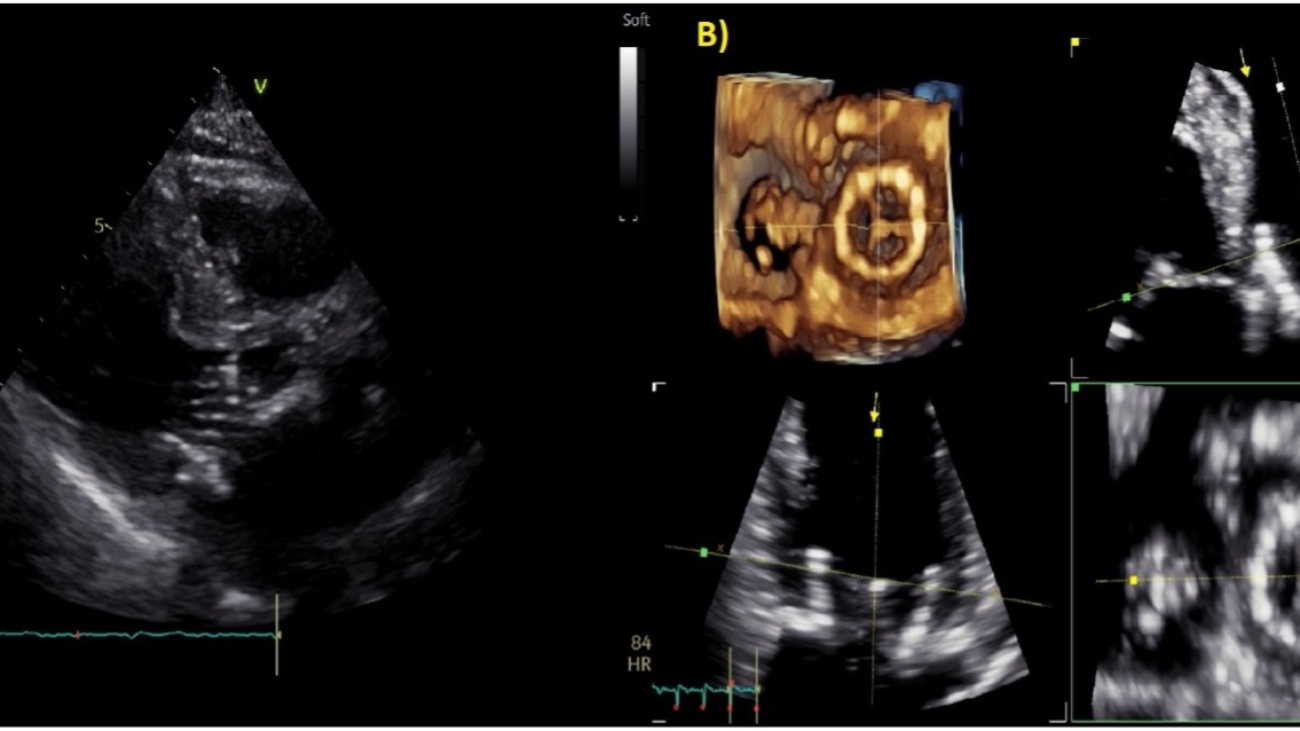
Posta l’indicazione alla procedura la paziente veniva sottoposta in prima istanza ad ecocardiogramma trans-esofageo (Figura 1) che evidenziava bioprotesi valvolare mitralica con evidenza di flail ascrivibile a lacerazione del lembo posteriore con insufficienza di grado severo.
Figura 1 – Insufficienza mitralica severa da degenerazione della bioprotesi mitralica. A) Ecocardiogramma trans-toracico. B) Ecocardiogramma trans-esofageo.
La paziente veniva quindi condotta in sala di Emodinamica dove veniva sottoposta ad anestesia generale ed intubazione oro-tracheale. Per via venosa femorale destra, si procedeva a puntura trans-settale sotto guida ecocardiografica transesofagea e successiva settoplastica. Veniva quindi eseguito impianto transcatetere di bioprotesi balloon-expandable Edwards-Sapien 3 29 mm in posizione mitralica durante pacing rapido a 160 bpm (Figura 2). La procedura decorreva in assenza di complicanze con buon risultato ecografico e fluoroscopico finale con minimo leak periprotesico.
Figura 2 – Impianto trans-catetere di protesi Edwards Sapien 3 in posizione mitralica. A) Rilascio del device balloon-expandable in corso di rapid pacing. B) Protesi in sede, normo-funzionante.
Al controllo ecocardiografico eseguito il giorno dopo la procedura si evidenziava protesi aortica in sede mitralica normofunzionante con lieve leak peri-protesico e normale gradiente trans-valvolare (2 mmHg); si segnalava minimo shunt sinistro-destro interatriale privo di effetto emodinamico.
Dopo una degenza decorsa in assenza di complicanze, la paziente veniva dimessa a distanza di tre giorni dalla procedura in terapia con Warfarin da proseguire per 3 mesi.
Al follow-up a 30 giorni la paziente si presentava in ambulatorio in ottime condizioni cliniche, asintomatica per angor, dispnea e palpitazioni riferendo un significativo miglioramento della qualità di vita. L’ecocardiogramma transtoracico (Figura 3), coadiuvato da valutazione strutturale 3D, evidenziava bioprotesi aortica in sede mitralica normofunzionante con normali gradienti trans-valvolari, invariato lo shunt sinistro-destro residuo. Si decideva quindi di proseguire la terapia in atto e si programmava successivo follow-up a tre mesi dalla procedura.
Figura 3 – Valutazione ecocardiografica trans-toracica della bioprotesi aortica in posizione mitralica a distanza di 30 giorni dall’impianto. A) Valutazione 2D in proiezione parasternale asse lungo. B) Valutazione 3D
Discussione
Negli ultimi anni c’è stato un significativo incremento dell’impianto delle bioprotesi valvolari rispetto alle valvole meccaniche per i ben noti vantaggi associati alle prime. Tuttavia, il principale limite delle bioprotesi è legato alla durata limitata nel tempo per la tendenza alla degenerazione strutturale. Si stima che la durata media di una bioprotesi in sede mitralica sia di 12 anni con un tasso di degenerazione del 2% a 5 anni dall’impianto con una rapidità di failure maggiore nei pazienti giovani ed affetti da insufficienza renale o epatica [1].
La disfunzione della bioprotesi valvolare viene distinta in quattro entità: degenerazione strutturale valvolare (SVD), degenerazione non strutturale valvolare (NVD), trombosi ed endocardite. La paziente del nostro caso clinico presentava una SVD, definita come variazioni permanenti intrinseche delle componenti tissutali della valvola, tra cui fissurazione e flail dei lembi protesici determinanti disfunzione che risulta nell’insorgenza di stenosi o insufficienza intra-protesica [2].
Secondo le linee guida della Società Europea di Cardiologia del 2021 riguardo il management delle valvulopatie, in caso di disfunzione di bioprotesi il re-intervento è raccomandato in pazienti sintomatici con un significativo incremento del gradiente intra-protesico, dopo aver escluso trombosi protesica, o insufficienza severa (Classe di Raccomandazione I, Livello di Evidenza C). Inoltre, l’impianto trans-catetere di protesi valve-in-valve in posizione mitralica e tricuspidalica può essere considerato in pazienti selezionati ad alto rischio di re-intervento chirurgico (Classe di Raccomandazione IIb, Livello di Evidenza B) [3].
L’impianto trans-catetere di bioprotesi aortica Edwards Sapien 3 in posizione mitralica, eseguito per la prima volta nel 2010 [4], è, ad oggi, l’unica strategia approvata per procedure valve-in-valve dedicate a pazienti con degenerazione di bioprotesi mitralica giudicati inoperabili chirurgicamente.
Il TMVR (Transcatheter Mitral Valve Replacement) registry è, attualmente, il più ampio registro sulla procedura di impianto trans-catetere di bioprotesi in sede mitralica. Tale studio ha incluso 521 pazienti sottoposti a TMVR per bioprotesi degenerata (valve-in-valve), anuloplastica fallita (valve-in-ring) o severa calcificazione dell’anulus mitralico (valve-in-mitral annular calcification) e ha mostrato che la procedura consente di raggiungere outcomes eccellenti per pazienti con bioprotesi degenerate nonostante l’elevato rischio chirurgico con un tasso di mortalità a 30 giorni del 6.2%, significativamente inferiore alla mortalità a 30 giorni riportata dopo re-intervento chirurgico (9.2-12.6%). Il registro ha altresì evidenziato che l’outcome più favorevole dell’intera coorte riguarda proprio i pazienti sottoposti ad impianto valve-in-valve. [5]
Dunque, l’impianto trans-catetere di bioprotesi aortica in sede mitralica rappresenta ad oggi non più soltanto un approccio promettente, ma un’importante strategia terapeutica con ogni probabilità destinata a diventare l’approccio di prima scelta per tali pazienti. Riteniamo fondamentale che in caso di degenerazione di bioprotesi la strategia terapeutica andrebbe personalizzata per ogni singolo paziente e discussa in Heart Team considerando il rischio chirurgico e le possibili difficoltà procedurali. Sottolineiamo quindi l’importanza di adattare l’approccio terapeutico alle sfide che i casi clinici impongono cercando e scegliendo, come nel nostro caso clinico, la protesi giusta al momento giusto.
Bibliografia
[1] Fann JI, Miller DC, Moore KA, Mitchell RS, Oyer PE, Stinson EB, Robbins RC, Reitz BA. Twenty-year clinical experience with porcine bioprostheses. Ann Thorac Surg.1996; 62:1301–1312.
[2] Capodanno D, Petronio AS, Prendergast B, et al. Standardized definitions of structural deterioration and valve failure in assessing long-term durability of transcatheter and surgical aortic bioprosthetic valves: a consensus statement from the European Association of Percutaneous Cardiovascular Interventions (EAPCI) endorsed by the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2017;38(45):3382-3390. doi:10.1093/eurheartj/ehx303
[3] 2021 ESC/EACTS Guidelines for the management of valvular heart disease. Vahanian A, et al. Eur Heart J. 2021 Aug 28;ehab395.
[4] Webb JG, Wood DA, Ye J, Gurvitch R, Masson JB, Rodes-Cabau J, et al. Transcatheter valve-in-valve implantation for failed bioprosthetic heart valves. Circulation 2010;121(16):1848-57.
[5] Yoon SH, Whisenant BK, Bleiziffer S, et al. Outcomes of transcatheter mitral valve replacement for degenerated bioprostheses, failed annuloplasty rings, and mitral annular calcification. Eur Heart J. 2019;40(5):441-451. doi:10.1093/eurheartj/ehy590
Basile Paolo, 1 Napoli Gianluigi 1, Tricarico Giuseppe 1, Falco Giorgia 1, Carella Maria Cristina 1, Anaclerio Matteo 1, Forleo Cinzia 1, Guaricci Andrea Igoren1.
1 U.O. Cardiologia Universitaria, Dipartimento di Emergenza e dei Trapianti d’Organo (DETO), Università di Bari “Aldo Moro”, Piazza G. Cesare 11, Bari (BA), 70124, Italy
ABSTRACT
La mutazione R222Q nel gene SCN5A è stata recentemente correlata alla sindrome MEPPC (multifocal ectopic Purkinje-related premature contractions) in cui, tramite un meccanismo di attività triggerata nelle fibre di Purkinje, la mutazione gain-of-function nel canale del sodio voltaggio-dipendente Nav1.5 determina extrasistoli atriali e ventricolari polimorfe e ripetitive con possibile evoluzione in cardiopatia dilatativa e/o morte improvvisa.
Il presente case report descrive un cluster familiare di sindrome MEPPC ad esordio con elevato burden aritmico non responsivo né ai beta-bloccanti né all’ablazione transcatetere dell’extrasistolia ventricolare. Il trattamento con flecainide ha invece determinato l’abolizione completa del fenotipo aritmico.
INTRODUZIONE
La sindrome MEPPC (multifocal ectopic Purkinje-related premature contractions) è stata recentemente associata alla mutazione autosomica dominante R222Q-SCN5A che, mediante un meccanismo di gain-of-function del gene SCN5A, determina una ripolarizzazione incompleta nelle cellule di Purkinje triggerando potenziali d’azione prematuri che si propagano ai ventricoli1, 2 con un elevato tasso di extrasistoli ventricolari polimorfe e tachicardie ventricolari non sostenute (TVNS).
Figura 1. Risonanza magnetica cardiaca con mezzo di contrasto che dimostra l’assenza anomalie strutturali cardiache. A, Asse corto. B, Quattro camere.
Descriviamo un cluster familiare di sindrome MEPPC esordita con un elevato burden aritmico in un paziente di 21 anni.
PRESENTAZIONE DEI CASI
Un paziente maschio di 21 anni è stato indirizzato presso il nostro centro per episodi sincopali non traumatici, a riposo, preceduti da sintomatologia neurovegetativa, con successivo riscontro all’ECG-Holter di periodi di ritmo giunzionale, idioventricolare e di blocco atrio-ventricolare di II° grado tipo 1 associati a TVNS polimorfe per le quali era stato avviato, con risultati trascurabili, trattamento con nadololo e flecainide a dosaggio ridotto (50 mg x2/die). Il paziente ha riferito familiarità per cardiomiopatia dilatativa (CMD) e morte cardiaca improvvisa (MCI), negando abitudine tabagica o uso di droghe. L’esame obiettivo, le indagini di laboratorio e l’ECG di ingresso sono risultati nella norma. Né l’ecocardiogramma né la risonanza magnetica (RMN) cardiaca hanno evidenziato anomalie strutturali (Figura 1).
(J, Complesso giunzionale; N, complesso sinusale; V, complesso ventricolare). Figura 2. ECG dinamico sec Holter delle 24 ore del padre del paziente. A, Ritmo giunzionale con dissociazione atrioventricolare isoritmica. B, Tachicardia ventricolare polimorfa non sostenuta.
Si è pertanto deciso di incrementare la posologia della flecainide (100 mg x2/die) e di procedere all’impianto di un loop-recorder. Nel follow-up a 6 mesi si è assistito alla completa remissione degli episodi aritmici. L’analisi genetica condotta durante il ricovero ha successivamente rivelato la mutazione R222Q-SCN5A, con successivo avvio dello screening genetico familiare e riscontro positivo nel padre.
L’uomo, di 56 anni, affetto da ipertensione arteriosa e dislipidemia mista, ha negato abitudini voluttuarie, sintomatologia anginosa, dispnea, cardiopalmo e/o sincopi. La valutazione iniziale è risultata anche in questo caso nella norma. L’ECG-Holter ha evidenziato periodi di ritmo giunzionale con dissociazione atrioventricolare isoritmica e un elevato numero di extrasistoli ventricolari polimorfe e TVNS provenienti principalmente dal tratto di efflusso del ventricolo destro (Figura 2), regredite durante sforzo e scarsamente responsive al metoprololo.
Per il riscontro ecocardiografico di lieve dilatazione e disfunzione sistolica del ventricolo sinistro, in assenza di fibrosi miocardica alla RMN cardiaca e di cause ischemiche alla coronarografia, si è optato per l’ablazione transcatetere dell’extrasistolia. Nonostante la regressione della disfunzione sistolica, l’efficacia dell’ablazione sul burden aritmico a 3 mesi è risultata trascurabile. Si è proceduto pertanto a testare la flecainide per via endovenosa (2 mg/Kg) con immediata regressione dell’extrasistolia ventricolare (Figura 3).
A 2 settimane dall’inizio della flecainide (100 mg x2/die), l’ECG-Holter ha confermato l’efficacia del farmaco (Tabella 1). Entrambi i pazienti sono attualmente in follow-up presso il nostro centro.
Tabella 1. ECG dinamico delle 24 ore del padre del paziente. Si può notare il netto decremento delle tachiaritmie sopraventricolari e ventricolari dopo assunzione della flecainide rispetto ai precedenti interventi terapeutici.
ExtrasistoliAtriali (n)
Extrasistoli Ventricolari (n)
TVNS (n)
Senza terapia
1315
7475
1
Con beta-bloccante
0
3776
1
Post-ablazione transcatetere
632
2496
127
Con flecainide
33
0
0
TVNS: tachicardia ventricolare non sostenuta.
DISCUSSIONE
Le mutazioni del gene SCN5A possono determinare un ampio spettro di sindromi: Brugada, QT lungo, CMD, e la rara sindrome MEPPC2-9. Quest’ultima è caratterizzata da una gain-of-function nel canale del sodio voltaggio-dipendente Nav1.510 che determina una ripolarizzazione incompleta nelle cellule di Purkinje generando potenziali d’azione prematuri che vengono condotti in via anterograda o retrograda a ventricoli o agli atri8 con un fenotipo clinico caratterizzato da extrasistoli ventricolari polimorfe ripetitive, tachiaritmie atriali, ritmo giunzionale, CMD potenzialmente reversibile e MCI. Non sono stati descritti né prolungamento dell’intervallo QT né sopraslivellamento del tratto ST2, 3, 5.
Il riscontro di tachiaritmie ventricolari necessita sempre una approfondita diagnosi differenziale. L’ECG da sforzo, eseguito nel padre, è un valido strumento diagnostico per escludere la tachicardia ventricolare polimorfa catecolaminergica. La coronaro-TC, la scintigrafia miocardica stress-rest o la coronarografia sono invece necessarie per escludere un’eziologia ischemica, specialmente nei pazienti con fattori di rischio cardiovascolare11. L’ablazione transcatetere è attualmente indicata nel sospetto di tachicardiomiopatia12, ma nel nostro caso non ha ottenuto gli effetti desiderati. Anche i beta-bloccanti, farmaci di prima linea nella gestione dell’extrasistolia, hanno sortito un modesto effetto terapeutico11. Dall’analisi della letteratura è emerso che i bloccanti dei canali del sodio come l’idrochinidina (classe IA) o la flecainide (classe IC), previa valutazione in regime ospedaliero della sicurezza e tollerabilità del farmaco, sono efficaci nel ridurre il burden aritmico e nel far regredire la disfunzione sistolica, quando presente2, 3, 5, 9. Il beneficio clinico sembrerebbe confermato anche al follow-up a lungo termine13. Attualmente non ci sono evidenze su un ruolo protettivo del defibrillatore impiantabile in questo contesto e ulteriori studi sono necessari per definire la gestione ottimale di questa rara sindrome.
Figura 3. Elettrocardiogramma del padre che mostra la remissione completa del ritmo giunzionale e dei complessi ventricolari prematuri dopo test farmacologico con flecainide e.v.
CONCLUSIONE
La presenza di numerose extrasistoli ventricolari e TVNS polimorfe, in presenza di familiarità per CMD e MCI, in un cuore strutturalmente normale o con segni di lieve disfunzione sistolica e dilatazione del ventricolo sinistro, in assenza di coronaropatia o fibrosi miocardica, può sollevare il sospetto della sindrome MEPPC. In tal caso il riscontro al test genetico della mutazione gain-of-function R222Q-SCN5A del canale Nav1.5 offre la conferma diagnostica e può indirizzare verso una terapia esclusivamente medica con flecainide. I beta bloccanti sono di dubbia utilità in questo contesto e l’ablazione transcatetere ha scarsi benefici a causa dei molteplici focolai di origine delle extrasistoli.
BIBLIOGRAFIA
1. Elliott, P.M., Multifocal ectopic Purkinje-related premature contractions: Sorting the wheat from the chaff. International Journal of Cardiology, 2018. 257: p. 218-219.
2. Laurent, G., et al., Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol, 2012. 60(2): p. 144-56.
3. Doisne, N., et al., A novel gain-of-function mutation in SCN5A responsible for multifocal ectopic Purkinje-related premature contractions. Hum Mutat, 2020. 41(4): p. 850-859.
4. Leventopoulos, G., et al., You cannot ablate the Lernaean Hydra: SCN5A mutation in a patient with multifocal ectopic Purkinje-related premature contractions syndrome treated with Flecainide and an implant of a subcutaneous defibrillator-a case report. Eur Heart J Case Rep, 2021. 5(4): p. ytab158.
5. Mann, S.A., et al., R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol, 2012. 60(16): p. 1566-73.
6. McNair, W.P., et al., SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation, 2004. 110(15): p. 2163-7.
7. Ter Bekke, R.M.A., et al., Beauty and the beat: A complicated case of multifocal ectopic Purkinje-related premature contractions. HeartRhythm Case Rep, 2018. 4(9): p. 429-433.
8. Wilde, A.A.M. and A.S. Amin, Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol, 2018. 4(5): p. 569-579.
9. Zakrzewska-Koperska, J., et al., Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: a case report. BMC Med Genet, 2018. 19(1): p. 94.
10. Remme, C.A., Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol, 2013. 591(17): p. 4099-116.
11. Priori, S.G., et al., 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J, 2015. 36(41): p. 2793-2867.
12. Pedersen, C.T., et al., EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Europace, 2014. 16(9): p. 1257-83.
13. Peters, S., et al., Long-Term Efficacy and Safety of Sodium Channel Antagonists in Patients With p.R222Q <i>SCN5A</i>-Related Arrhythmic Dilated Cardiomyopathy. JACC: Clinical Electrophysiology, 2021. 7(1): p. 126-128.
Andrea Angelozzia, Leonardo Belfiorettia, Marco Marinib, Tommaso Pivab, Francesca Patanib, Antonio Dello Russoa, Gian Piero Pernab, Marco Di Eusanioc
a Clinica di Cardiologia e Aritmologia, Dipartimento di Scienze Cardiovascolari, Azienda Ospedaliero-Universitaria, Ospedali Riuniti di Ancona
b S.O.D. Cardiologia-Emodinamica-UTIC, Dipartimento di Scienze Cardiovascolari, Azienda Ospedaliero-Universitaria, Ospedali Riuniti di Ancona
c S.O.D. Cardiochirurgia, Ospedali Riuniti “Umberto I-Lancisi-Salesi”, Dipartimento di Medicina Clinica e Sperimentale, Università Politecnica delle Marche, Ancona
Abstract
La rottura ischemica del muscolo papillare con sviluppo di insufficienza mitralica acuta emodinamicamente significativa è una delle complicanze meccaniche più temute degli infarti miocardici STEMI. Nonostante la riduzione dell’incidenza negli ultimi anni, questa condizione è gravata ancora oggi da una mortalità estremamente elevata. In questo report presentiamo il caso di una paziente con rottura ischemica di muscolo papillare determinante insufficienza mitralica severa complicata da edema polmonare acuto trattata mediante impianto di MitraClip, dato il rischio proibitivo dell’intervento cardiochirurgico convenzionale.
Caso clinico
Una donna di 85 anni si presentava presso il nostro Pronto Soccorso per dispnea al minimo sforzo, comparsa da circa un giorno, in assenza di dolore toracico. In anamnesi una nota cardiopatia ischemica cronica sottoposta circa 10 anni prima a procedura di rivascolarizzazione miocardica chirurgica mediante CABG (arteria mammaria interna di sinistra su interventricolare anteriore e vena safena in sequenziale su interventricolare posteriore e ramo marginale ottuso), DM tipo II, ipertensione arteriosa sistemica e dislipidemia.
La paziente all’ingresso presentava stabilità emodinamica con valori di PA pari a 150/80 mmHg e all’ECG veniva documentato sopraslivellamento del tratto ST come da infarto transmurale infero-postero-laterale. L’ecoscopia eseguita in urgenza mostrava una severa riduzione della funzione sistolica ventricolare sinistra (FEVS 30-35%) per acinesia del segmento medio-basale della parete infero-posteriore e dell’apice inferiore, un ventricolo destro di normali dimensioni e normocinetico, una vena cava inferiore dilatata e normocollassante con gli atti respiratori mentre dal punto di vista valvolare evidenziava una insufficienza mitralica di grado moderato-severo. Agli esami ematochimici elevazione dei marker specifici di miocardionecrosi (Troponina I hs 8.031 ng/L) e dei peptidi natriuretici (BNP 2209 ng/L).
Figura 1 – Insufficienza mitralica massiva pre Mitraclip
La paziente veniva trasferita in sala di emodinamica per esecuzione di studio coronarografico urgente.
All’arrivo in sala di emodinamica si verificava peggioramento della dispnea con sviluppo di edema polmonare acuto e ortopnea obbligata. Dopo aver stabilizzato la paziente mediante C-PAP, terapia diuretica endovenosa e nitrato in infusione continua si procedeva a studio coronarografico urgente che mostrava pervietà dell’AMIS per IVA e occlusione del graft venoso dopo l’anastomosi con IVP con evidenza di estesa occupazione trombotica. Veniva quindi eseguito tentativo di ricanalizzazione meccanica e dilatazione transluminale di arteria circonflessa al passaggio fra primo e secondo segmento, risultato tuttavia inefficace e successivamente si procedeva a ricanalizzazione meccanica e dilatazione transluminale del graft venoso per IVP-MO in corrispondenza dell’interponte, a valle dell’anastomosi con IVP, mediante POBA e tromboaspirazione manuale, risultato efficace nel ripristino della pervietà del bypass, pur in presenza di flusso rallentato.
La paziente veniva poi trasferita in UTIC dove giungeva tachipnoica, oligurica e lievemente disorientata. All’EGA si documentava quadro di acidosi metabolica con severa insufficienza respiratoria (pH 7,23, P/F 136, lattati 12 mmol/l) per cui veniva posizionato casco C-PAP.
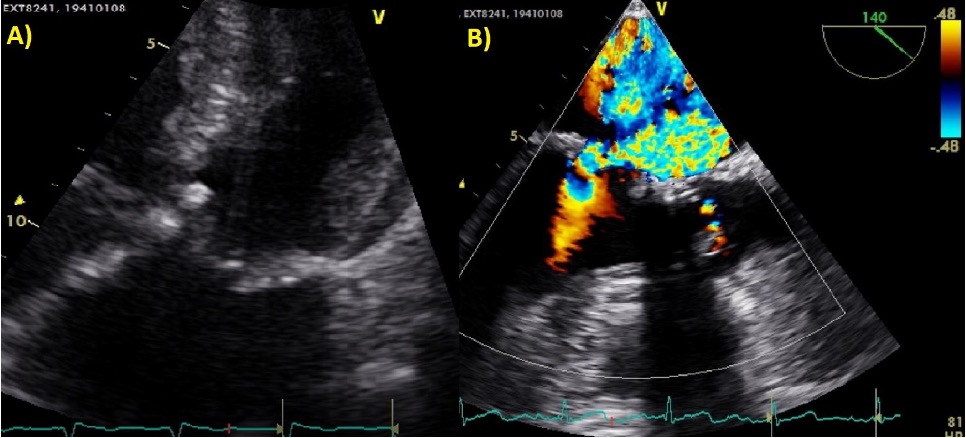
All’ecocardiogramma eseguito a letto della paziente si evidenziava rottura parziale del muscolo papillare postero-mediale a livello della testa determinante un’insufficienza mitralica di grado severo con jet eccentrico diretto postero-inferiormente e con estensione intercommissurale > 15 mm.
Effettuata la valutazione cardiochirurgica, si controindicava l’intervento per rischio operatorio proibitivo, in quanto già in esiti di precedente sternotomia, età avanzata e quadro acuto con severa disfunzione ventricolare sinistra. Si optava pertanto per tentativo di stabilizzazione del quadro clinico mediante terapia farmacologica e supporto meccanico al circolo mediante impianto di contropulsatore aortico (IABP). Inoltre, data la persistenza di oligo-anuria, si rendeva necessaria avviare terapia sostitutiva renale mediante CVVHD in calcio-citrato.
Le manovre terapeutiche effettuate, unite al supporto ventilatorio non invasivo con C-PAP, hanno consentito una progressiva riduzione dei lattati con normalizzazione del pH, un miglioramento degli scambi gassosi e una netta riduzione dello stato di congestione polmonare. Tuttavia la paziente risultava “IABP dipendente”, con diversi tentativi di weaning falliti. Per tale motivo, alla luce dell’inoperabilità della paziente, veniva sottoposta ad ETE per l’eventuale candidabilità a Mitraclip.
All’ETE si documentava la presenza dei criteri anatomici per procedere con tale procedura per cui si procedeva pertanto a posizionamento di due MitraClip (1 clip NTR + 1 clip XTR) in anestesia generale. Al rientro in UTIC veniva ripetuto un ecocardiogramma TT che mostrava una insufficienza valvolare residua di grado lieve-moderato in presenza di gradiente medio all’afflusso ventricolare sinistro non significativo (pari a 3 mmHg). Nelle ore successive l’impianto i parametri emogasanalitici evidenziavano buoni scambi respiratori (P/F 350) e lattati nei limiti di norma per cui la paziente veniva estubata. Successivamente si rendeva possibile la riduzione del supporto farmacologico e meccanico con lo IABP, il quale veniva rimosso il giorno successivo. Si assisteva inoltre ad un progressivo miglioramento della funzionalità renale e al ripirsitono di valida diuresi spontanea per cui veniva sospesa CVVHD. La paziente veniva infine dimessa 5 giorni dopo la procedura.
Discussione
Figura 2 – Insufficienza mitralica post impianto Mitraclip
Tra le complicanze meccaniche più temute degli infarti transmurali c’è sicuramente la rottura di muscolo papillare che, quando non trattata, ha una mortalità del 50% nelle prime 24h e più dell’80% a una settimana [1][2]. Tale complicanza si verifica dall’1% al 5% dei pazienti con infarto miocardico acuto ed è causa del 5% circa dei decessi infarto-correlati [3]. Il muscolo papillare postero-mediale è più frequentemente coinvolto poiché, al contrario del muscolo papillare antero-laterale, non ha una doppia irrorazione coronarica. Il trattamento di scelta di questa condizione è, nei casi in cui è possibile, la sostituzione valvolare mitralica o la riparazione valvolare mediante cardiochirurgia. Tuttavia è estremamente frequente che questi pazienti presentino un rischio operatorio estremamente elevato, tale da rendere proibitivo qualunque approccio “invasivo” mediante cardiochirurgia. La riparazione percutanea della valvola mitralica mediante MitraClip può rappresentare una valida possibilità in acuto e il nostro caso clinico suggerisce che questa tecnica minimamente invasiva può essere utile per pazienti ad alto rischio con shock cardiogeno ed edema polmonare dopo IMA complicato da insufficienza mitralica severa.
Bibliografia
[1] Bouma W, Wijdh-den Hamer IJ, Klinkenberg TJ, Kuijpers M, Bijleveld A, van der Horst ICC, Erasmus ME, Gorman JH, Gorman RC, Mariani MA.. Mitral valve repair for post-myocardial infarction papillary muscle rupture. Eur J Cardiothorac Surg 2013;44:1063–1069.
Scuola di Specializzazione in Malattie dell’Apparato Cardiovascolare, Milano Bicocca
ABSTRACT
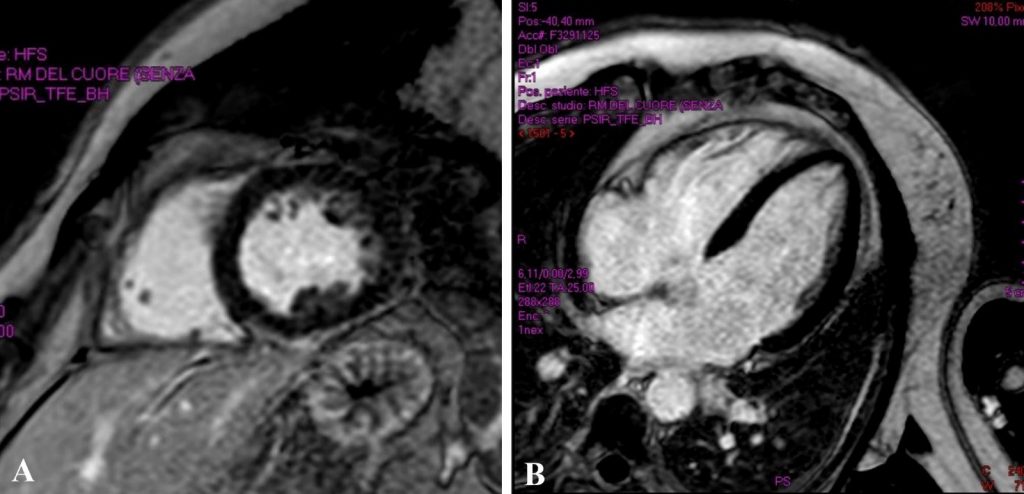
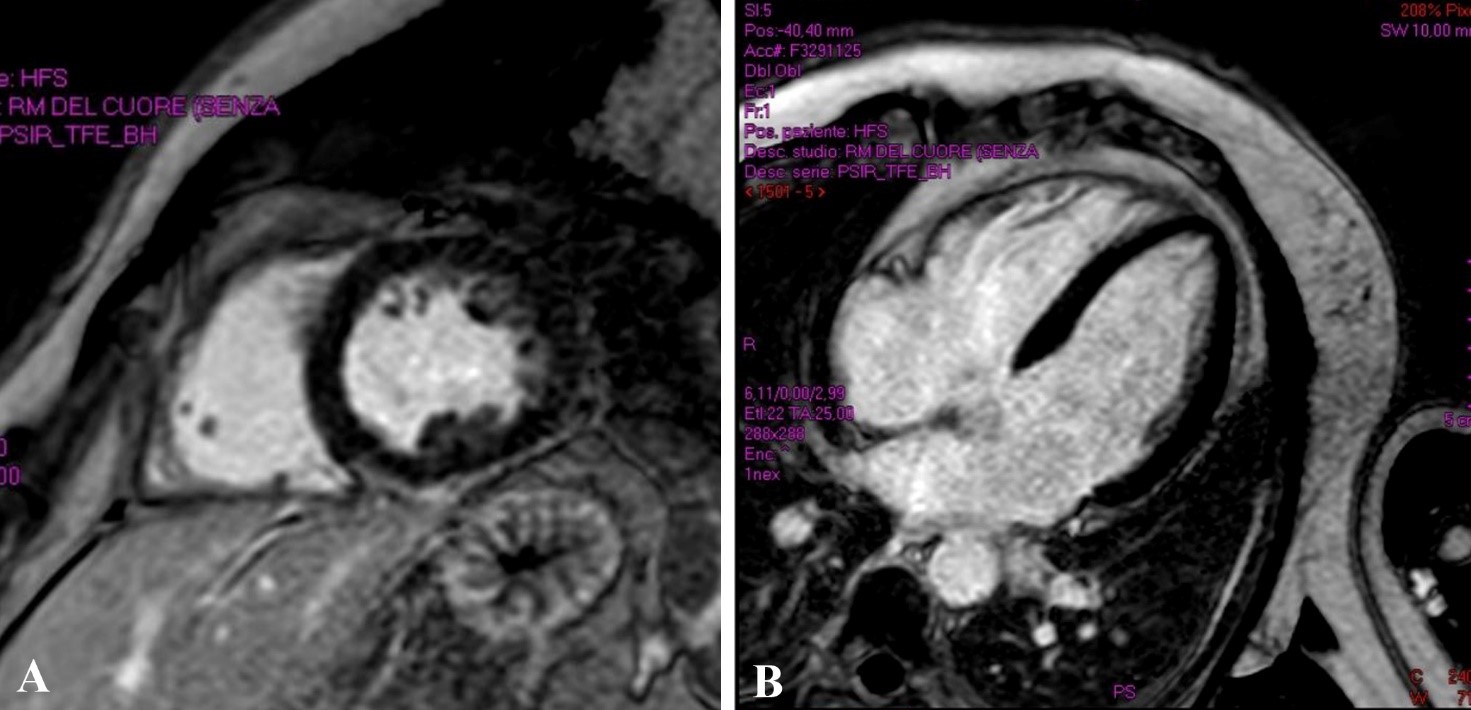
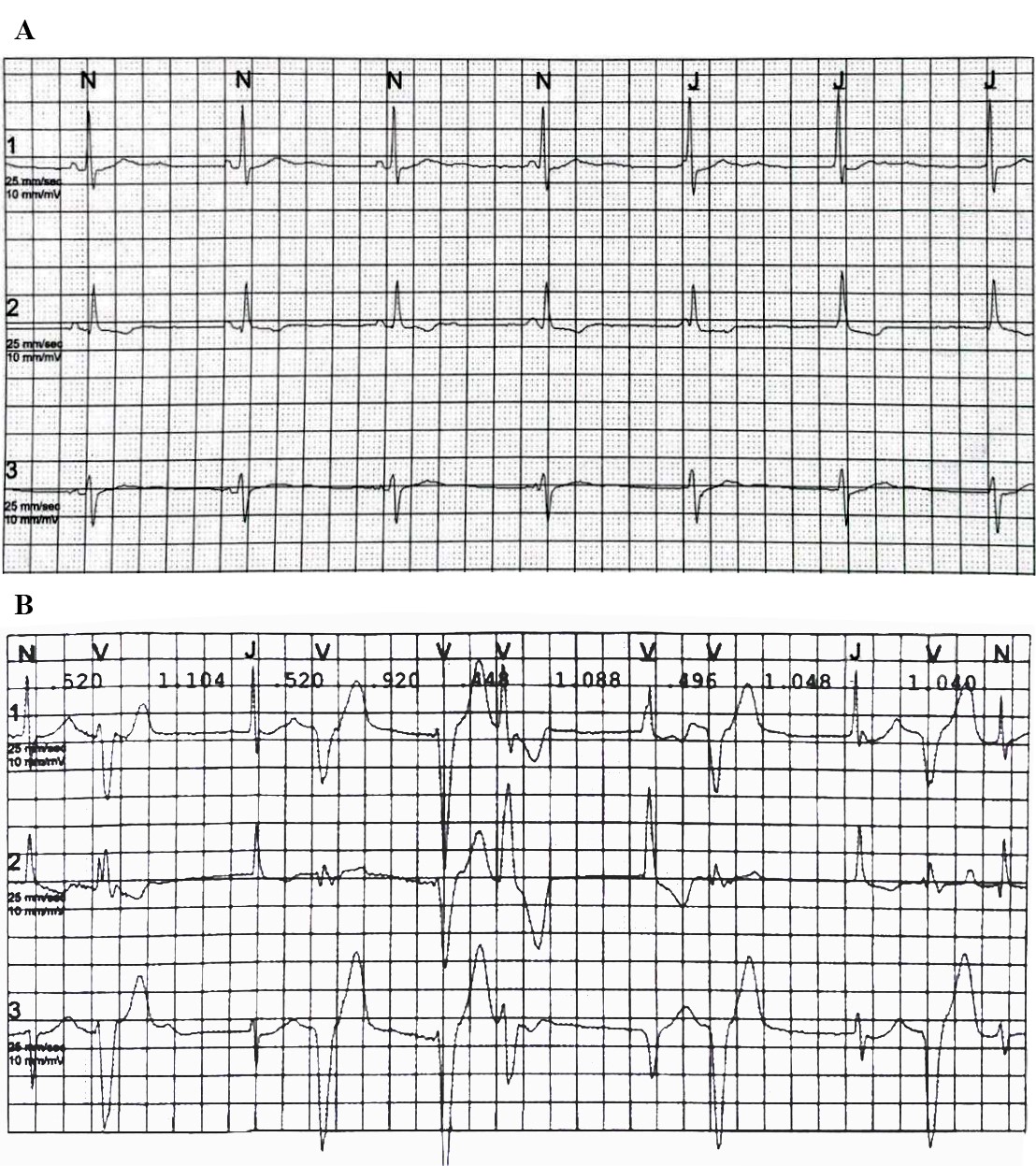
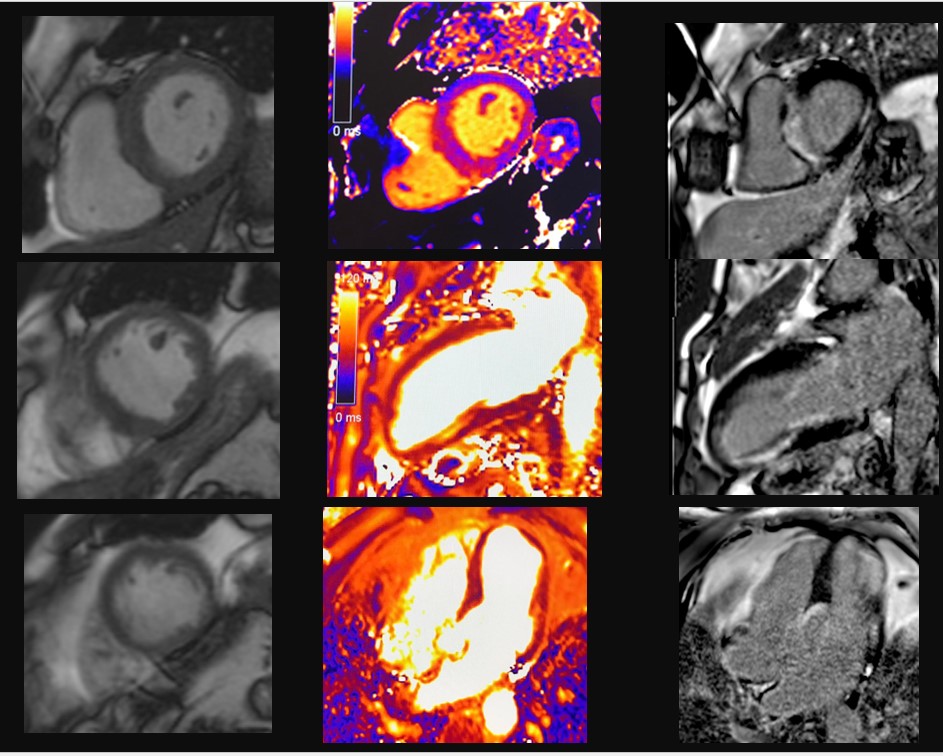
Un paziente di 77 anni viene ricoverato per accertamenti in seguito a riscontro di frequenti episodi di tachicardia ventricolare non sostenuta (TVNS). In anamnesi riporta ipertensione arteriosa, diabete mellito e sarcoidosi polmonare in remissione da più di dieci anni. Alla telemetria (TLM) vengono registrate frequenti TVNS a più morfologie. La risonanza magnetica (RM) cardiaca evidenzia presenza di late gadolinium enhancement (LGE) infero-postero-laterale e settale con edema alle sequenze di mapping. Dato l’alto sospetto clinico viene iniziata la terapia per sarcoidosi cardiaca con cortisone ad alte dosi e beta bloccante, ottenendo una drastica riduzione del burden aritmico. La tomografia ad emissione di positroni (PET) cardiaca, tuttavia, risulta essere negativa. Vengono discusse due possibili spiegazioni della discrepanza fra reperti RM e PET in corso di sarcoidosi cardiaca.
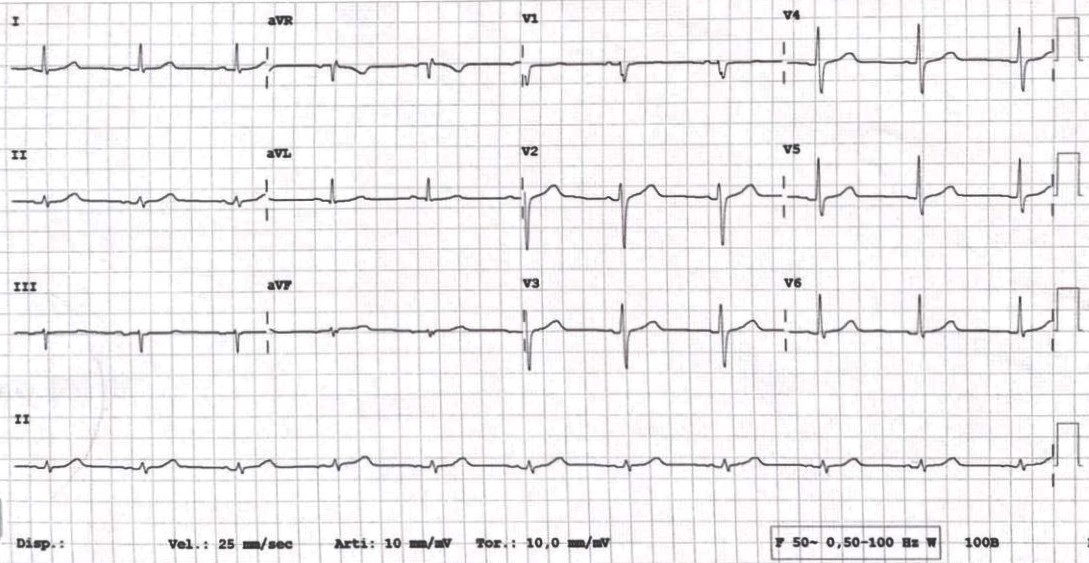
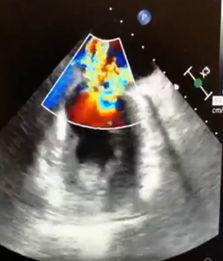
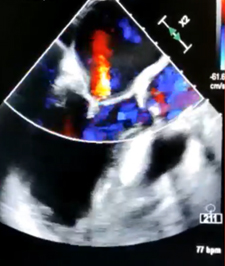
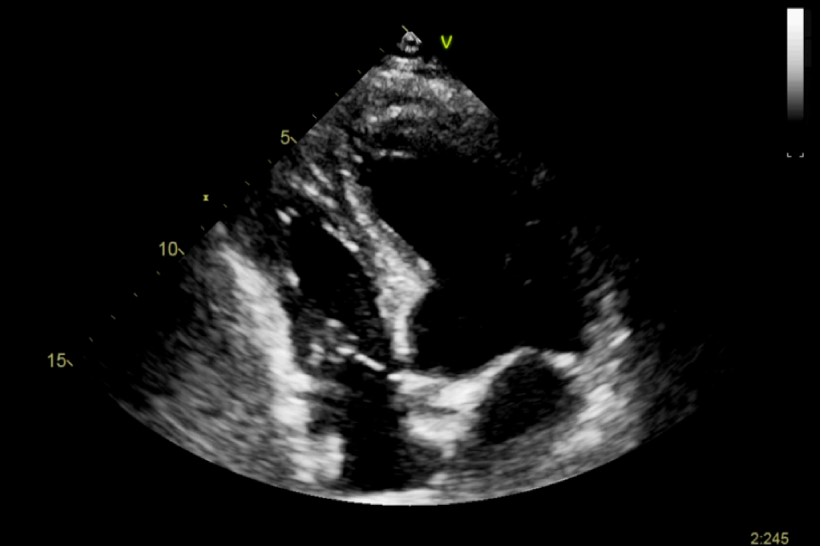
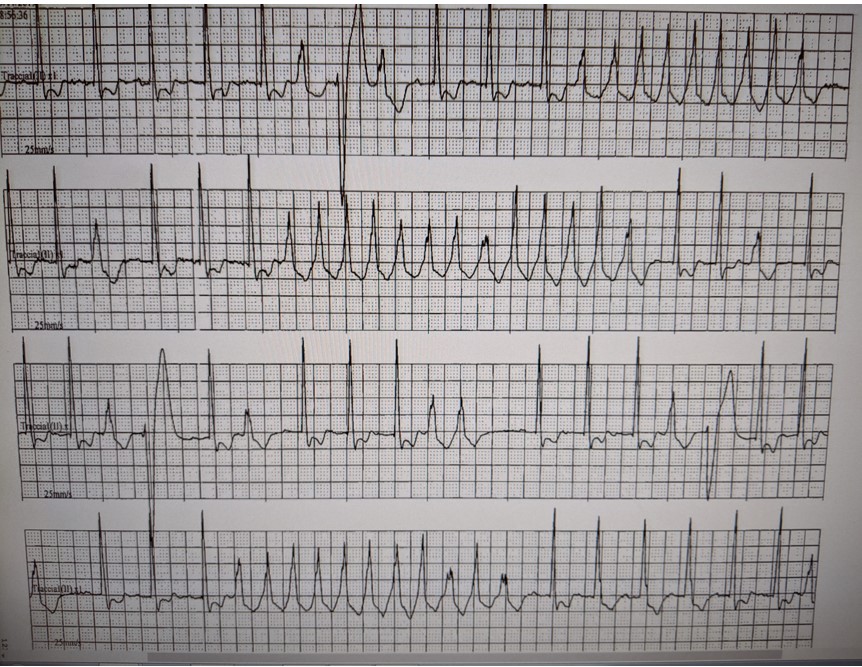
CASO CLINICO
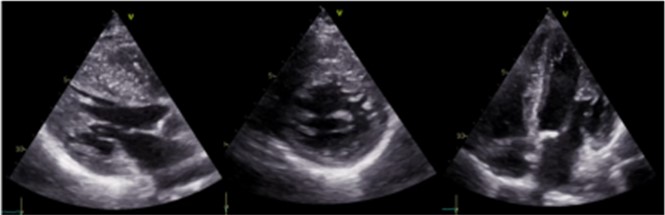
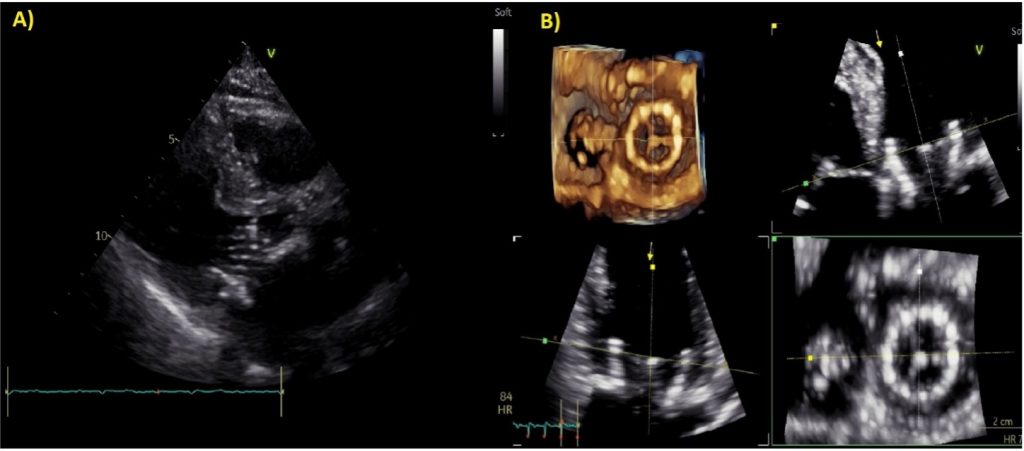
Figura 1. Ecocardiogramma con proiezione 5 camere.
Un paziente di 77 anni giungeva in ambulatorio portando in visione il monitoraggio elettrocardiografico di 24 ore secondo Holter, eseguito per episodi pre-sincopali, che evidenziava fasi di blocchi atrio-ventricolari (BAV) di secondo grado tipo Mobitz I, frequentissimi battiti extrasistolici ventricolari (BEV) e numerose salve di TVNS. Il paziente riferiva l’esecuzione di un Holter ECG due anni prima, portato in visione all’equipe medica, che evidenziava rare salve di TVNS, per cui aveva avviato la terapia con amiodarone, in assenza di beneficio clinico. In tale occasione tra gli accertamenti aveva eseguito uno studio coronarografico, risultato negativo. In anamnesi riportava ipertensione arteriosa, in terapia con diuretico e calcio antagonista, e diabete mellito tipo 2 in terapia con ipoglicemizzante orale. All’età di 50 anni aveva ricevuto una diagnosi istologica di sarcoidosi polmonare trattata con terapia steroidea e da allora in remissione.
In considerazione della sintomaticità e della frequenza degli eventi aritmici ventricolari il paziente veniva ricoverato per ulteriori accertamenti.
Obiettivamente all’ingresso il paziente era in buon compenso cardiocircolatorio. L’ECG a 12 derivazioni mostrava un ritmo sinusale normofrequente condotto con BAV I, blocco di branca destra (BBDx) ed emiblocco anteriore sinistro (EAS). Eseguiva quindi ecocardiogramma transtoracico che rilevava un assottigliamento del setto interventricolare (Figura 1) prossimale con movimento asincrono setto-apicale, frazione d’eiezione (FE) del 55%, pressione polmonare arteriosa sistolica (PAPs) 40-50 mmHg e insufficienza mitralica e tricuspidale di grado moderato.
Alla telemetria ECG vengono rilevate frequenti salve di TVNS con morfologia variabile (Figura 2).
Figura 2. Tracciato telemetrico ECG con la registrazione di frequenti tachicardie ventricolari non sostenute monomorfe.
Veniva quindi eseguita, durante il ricovero, la RM cardiaca che rilevava edema della parete infero-postero-laterale medio-distale e del setto in toto, nelle sequenze T1 e T2 mapping, e LGE con pattern intramiocardico (non ischemico) nelle medesime regioni (Figura 3).
In considerazione dell’elevato sospetto clinico di coinvolgimento cardiaco di sarcoidosi sistemica, veniva iniziata terapia con cortisone ad alte dosi (prednisone 37,5 mg) e betabloccante (carvedilolo 6,25 mg x 2). Il paziente veniva inoltre sottoposto a impianto di un defibrillatore impiantabile (ICD) bicamerale.
Per confermare la diagnosi, il paziente veniva inviato ad eseguire una PET con 18F-FDG che, tuttavia, risultava negativa per uptake patologico nel tessuto miocardico.
Nonostante l’esito inaspettatamente negativo della PET, veniva proseguita la terapia farmacologica già impostata. A 48 ore dall’inizio della terapia si assisteva ad una drastica riduzione del burden aritmico alla telemetria, per cui il paziente veniva dimesso al domicilio. Alla visita di follow-up dopo 3 mesi il paziente riferiva un ottimo controllo dei sintomi e nessuna scarica dell’ICD è stata registrata.
DISCUSSIONE
Abbiamo presentato un caso di sarcoidosi cardiaca con una inusuale discrepanza fra reperti RM e PET.
Figura 3. Immagini di RM cardiaca con sequenze T1, T2 e acquisizione di LGE.
La sarcoidosi cardiaca si manifesta nel 5-25% dei pazienti affetti da sarcoidosi con blocchi di conduzione, aritmie ventricolari e scompenso cardiaco. La diagnosi viene posta in seguito a conferma istologico o, più frequentemente, secondo dei criteri diagnostici basati sulla compresenza di: 1) conferma istologica di sarcoidosi extracardiaca; 2) almeno uno fra cardiomiopatia responsiva a terapia steroidea, FE<40% altrimenti inspiegata, presenza di TV altrimenti inspiegate, BAV II Mobitz II o BAV III, uptake patologico alla PET, LGE alla RM cardiaca, uptake patologico di gallio; 3) assenza di altre cause che spieghino le manifestazioni cardiache del paziente (1).
I due esami di imaging più frequentemente utilizzati sono la PET e la RM cardiaca. Ciascuno di questi esami ha una accuratezza riportata del 75-90% e, secondo i criteri diagnostici, possono essere usati alternativamente per confermare la diagnosi di sarcoidosi cardiaca.
Una prima ipotesi avanzata per giustificare la discrepanza fra reperti RM e PET è stata una possibile inadeguata preparazione all’esame di PET cardiaca. E’ infatti noto come il metabolismo miocardico sia glucidico esclusivamente in presenza di insulina e, in assenza di questo ormone sia basato sul metabolismo degli acidi grassi. L’obiettivo della preparazione è quindi di abbattere i livelli di insulina tramite una dieta ricca in acidi grassi e povera in glucosio nelle 48 ore precedenti all’esame (oltre all’astinenza dall’esercizio fisico e dalla somministrazione di insulina). Un’inadeguata osservanza di questa preparazione porta quindi il paziente ad eseguire l’esame con l’intero miocardio, e non solo con l’infiltrato cellulare patologico, dipendente dal metabolismo glucidico e quindi captante 18-FDG. L’assenza di un gradiente di captazione fra miocardio sano ed infiltrato determina quindi una considerevole riduzione della sensibilità dell’esame. Tuttavia, nel nostro caso, tutti i criteri di preparazione sono stati osservati.
Un’ulteriore ipotesi, che è stata avanzata, si basa sulle differenti alterazioni istologiche che i due esami sono in grado di evidenziare: nella RM il gadolinio tende ad accumularsi nella matrice extracellulare, che risulta essere aumentata sia in caso di infiltrato edematoso (fase acuta della malattia) che fibroso (esiti della malattia). La distinzione fra le due fasi viene poi effettuata tramite ulteriori sequenze, quali le T1 e T2 mapping, che sono specifiche nel riconoscere l’edema. D’altro canto, nella PET il 18-FDG viene captato specificamente dall’infiltrato cellulare, tipico della fase acuta della malattia. Si può quindi ipotizzare che il nostro paziente avesse un infiltrato flogistico di fase acuta prevalentemente edematoso, per cui LGE evidenziato dall’indagine RM, e viceversa con scarsa componente cellulare, per cui PET negativo. Questa ipotesi, che ad oggi non trova riscontro in letteratura, potrebbe essere spunto per futuri studi volti a individuare differenze nella sensibilità di RM e PET cardiaca nel riconoscere la fase acuta della sarcoidosi cardiaca.