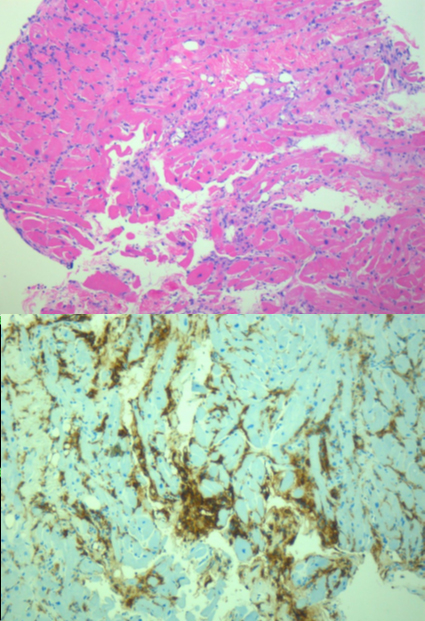
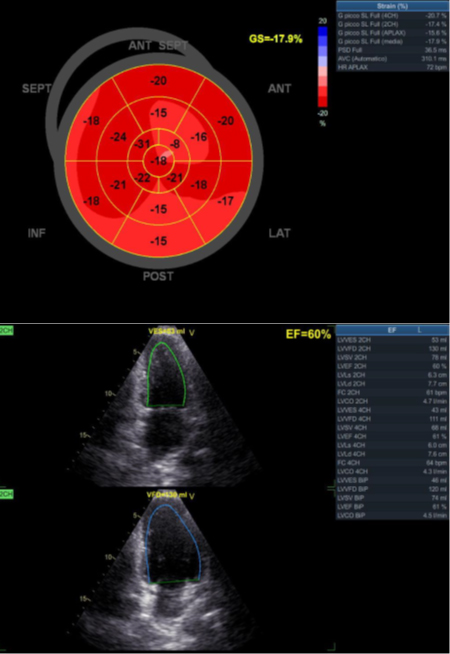
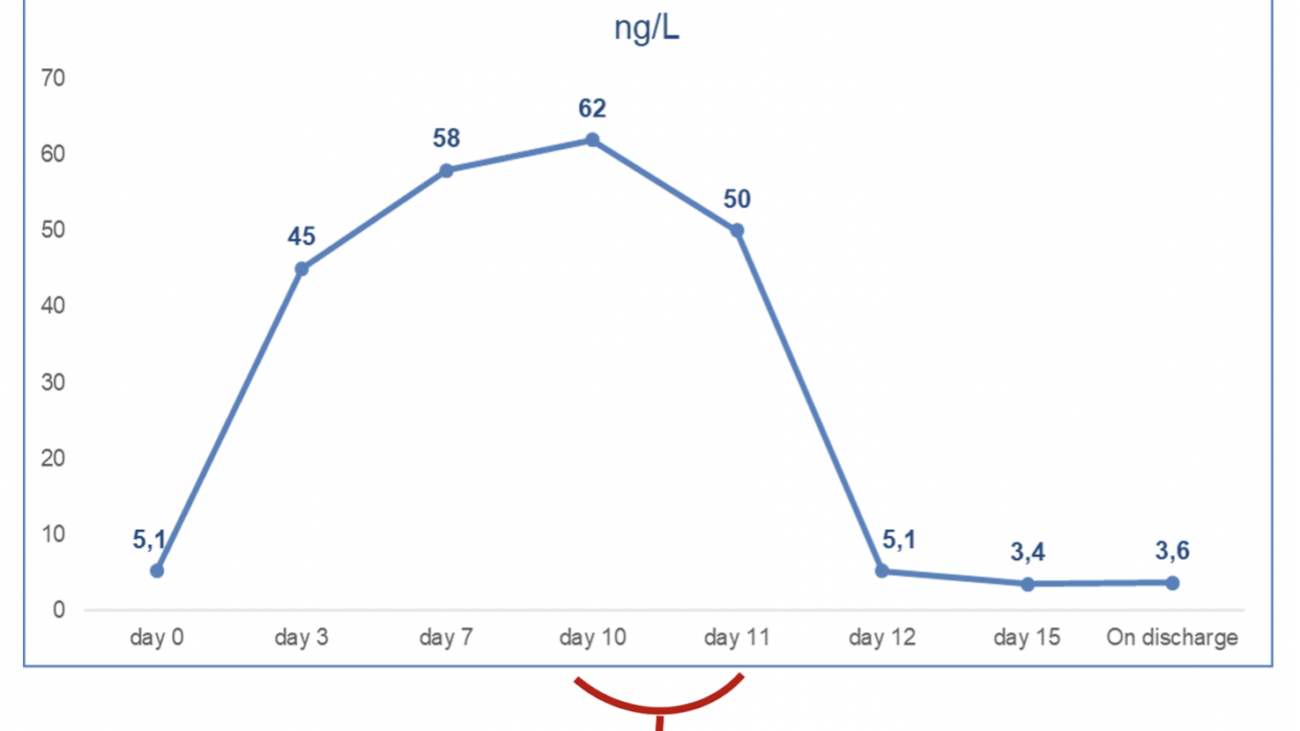
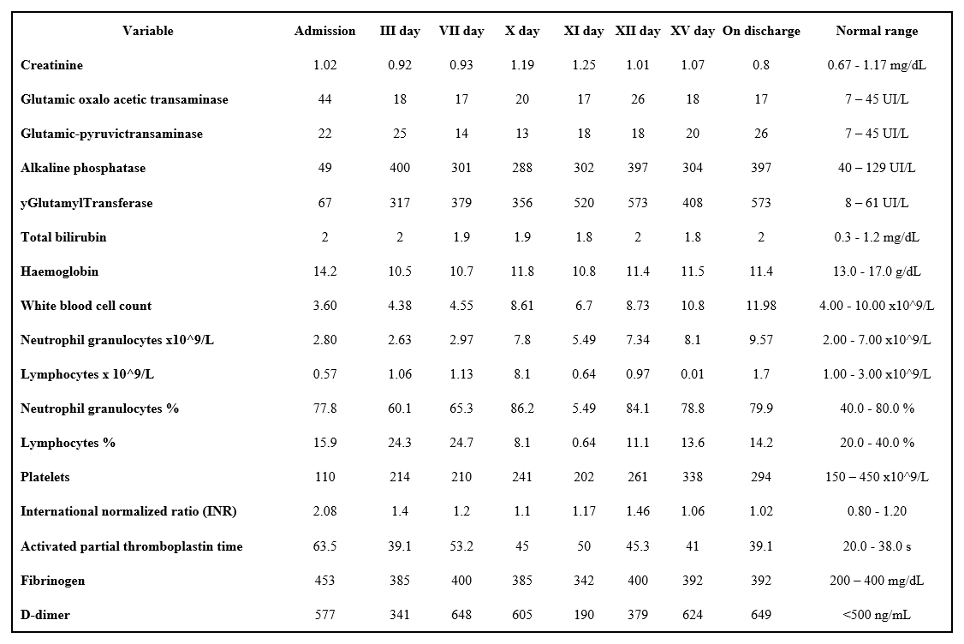
Francesca Maria Di Muro, MD, Miroslava Stolcova, MD, Alessio Mattesini MD, Giulia Nardi MD, Niccolò Ciardetti MD, MD, Carlo Di Mario, MD, PhD, FESC, FACC, FSCAI, FRCP, Francesco Meucci, MD.
Structural Interventional Cardiology, Department of Clinical and Experimental Medicine, Careggi University Hospital, Clinica Medica, Room 124, Largo Brambilla 3, 50134 Florence, Italy
Abstract
La chiusura percutanea dell’appendice atriale sinistra rappresenta una alternativa terapeutica per la prevenzione del rischio cardioembolico nei pazienti con fibrillazione atriale non valvolare ad elevato rischio emorragico. Lo studio preprocedurale della morfologia e delle dimensioni auricolari rappresentano uno step chiave per garantire un impianto di successo. Il nostro caso clinico descrive l’utilizzo di un programma di simulazione 3D (FEops NV, Ghent, Belgium) applicato allo studio di un paziente con auricola sinistra con morfologia atipica (a coda di balena) che ha permesso un corretto posizionamento di sistema Watchman.
Introduzione
La chiusura percutanea dell’appendice atriale sinistra è una procedura efficace nella prevenzione degli eventi cardioembolici e dello stroke ischemico in caso di fibrillazione atriale non valvolare ed è una valida alternativa all’anticoagulazione nei pazienti con storia di sanguinamento1. Sebbene siano riconosciute diverse morfologie di auricola sinistra, ne sono state identificate 4 prevalenti: cactus, windsock (o a manica di vento), cavolfiore e chicken wing (ala di pollo)2. Le conformazioni che non ricadono in queste categorie richiedono un’analisi pre-procedurale più accurata per ottenere una corretta selezione del device e un impianto ottimale.
Questo case-report illustra l’importanza di un programma di simulazione 3D, sviluppato da FEops (FEops NV, Ghent, Belgium) basato sulle immagini TC, che permette di selezionare la proiezione di lavoro e il tipo di device da impiantare, predicendone la posizione e il grado di compressione finale.3
Caso clinico
Un uomo di 62 anni con storia di fibrillazione atriale permanente (CHA2DS2-VASc = 2) in terapia con Warfarin viene riferito al nostro centro per sottoporsi a chiusura di auricola sinistra a seguito di sviluppo di ematoma cerebellare nel Dicembre 2020. Da segnalare in anamnesi adenocarcinoma uroteliale di basso grado sottoposto ad intervento di resezione transuretrale di tumore della vescica (TURBT) ed un recente intervento chirurgico di rimozione di adenocarcinoma polmonare localizzato.
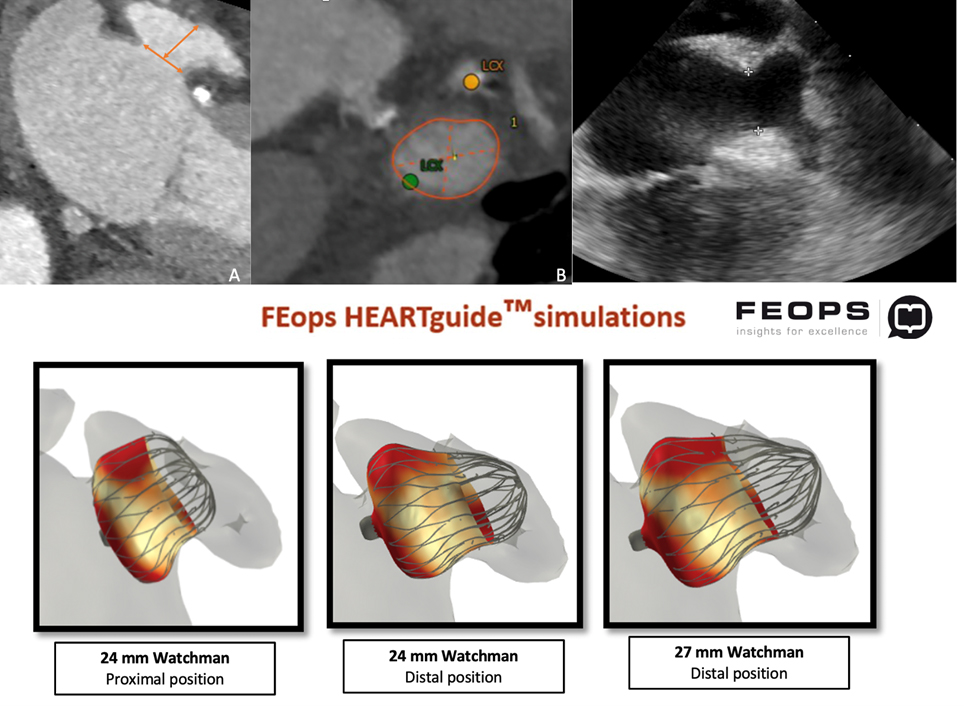
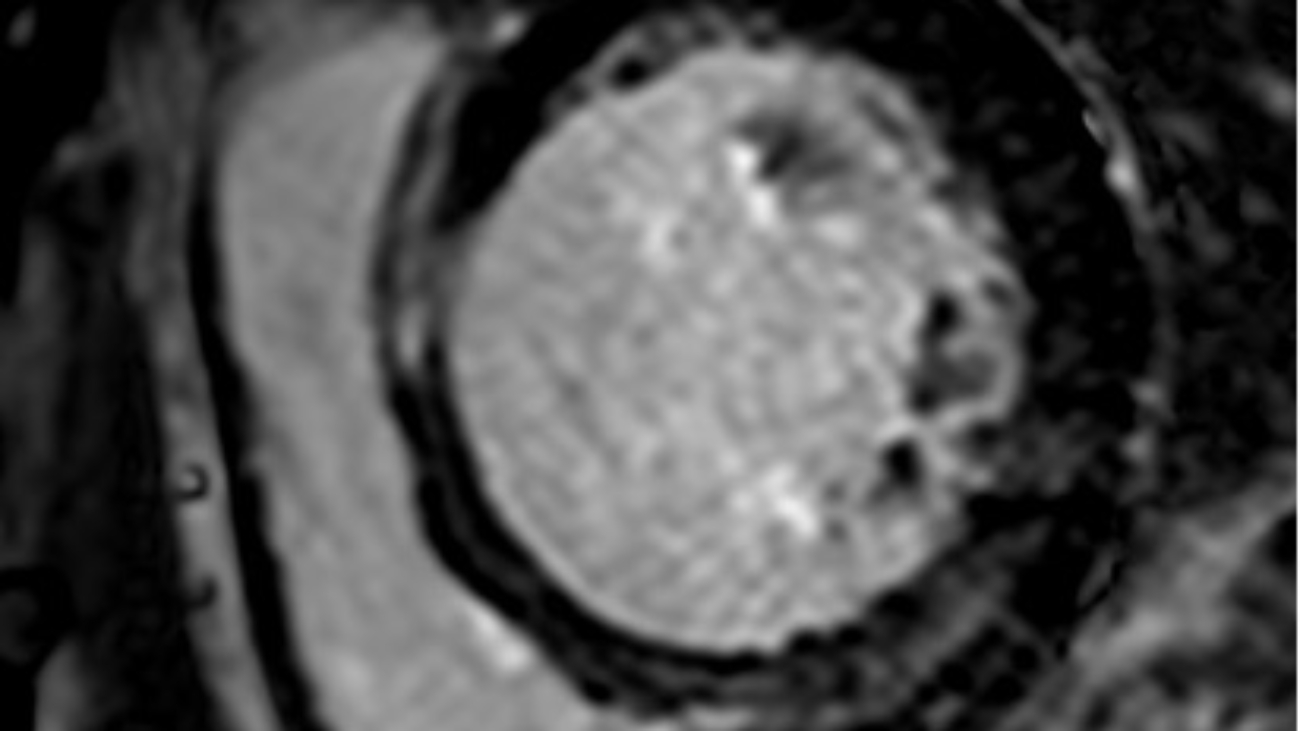
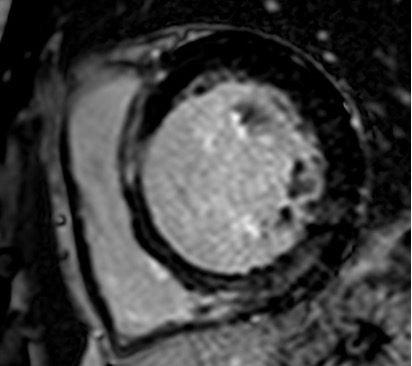
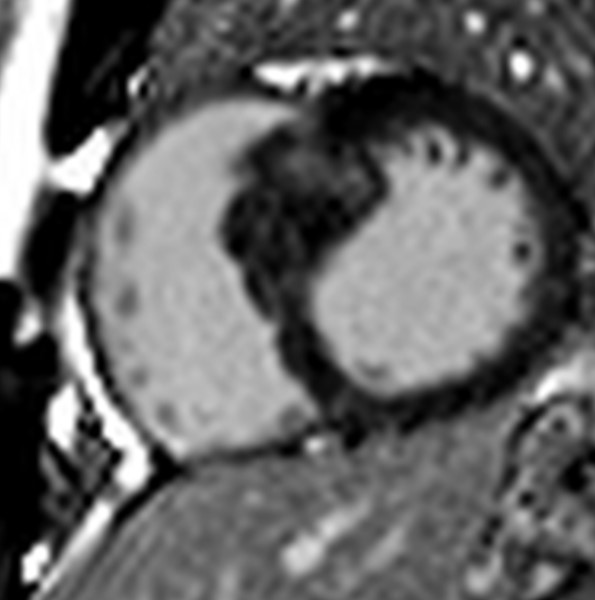
La TC pre-procedurale mostrava una anatomia dell’auricola estremamente sfavorevole, con un breve collo e due lobi prossimali simmetrici opposti e speculari l’uno all’altro, misurando una landing zone di 16 x 22 mm ed una profondità di impianto pari a 12 mm.
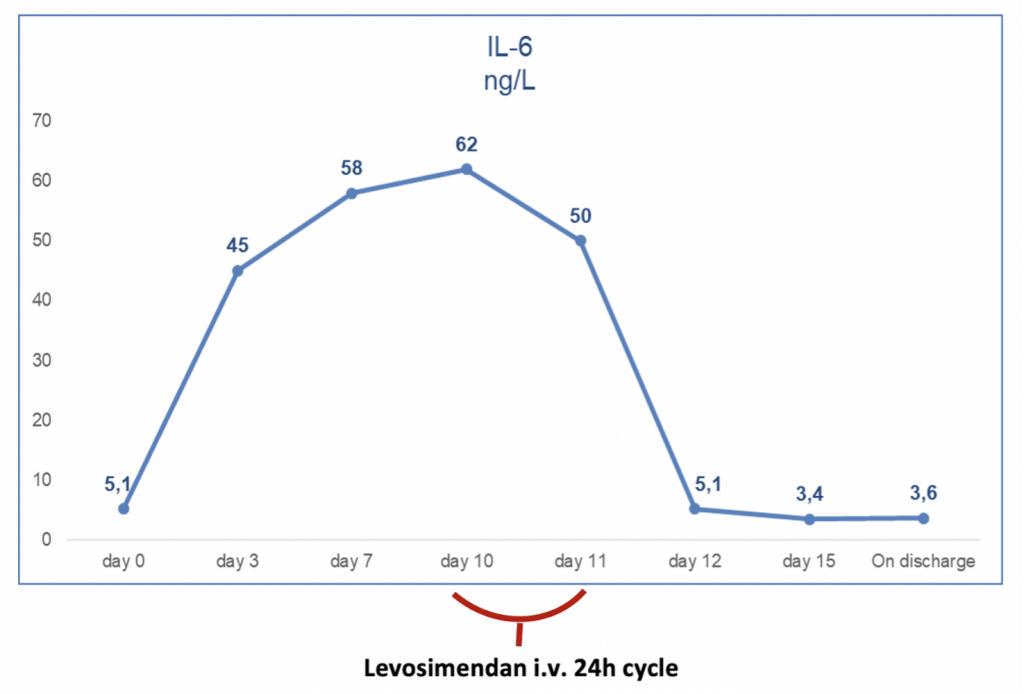
Vista questa rara anatomia, altresì nota come auricola a coda di balena, abbiamo deciso di ottenere una predizione dell’impianto mediante il supporto della piattaforma online sviluppata da FEops HEARTguide. Il software FEOps è in grado di simulare device di varie dimensioni e diverse profondità di impianto, proponendo il modello pre-operatorio più adatto all’anatomia riscontrata tramite imaging. Nel nostro caso FEOps ha suggerito l’impianto prossimale di un dispositivo Watchman 24 mm con una compressione finale predetta di circa il 10%, un buon grado di apposizione in assenza di eccessiva protrusione in atrio sinistro (Figura 1).
La procedura è stata eseguita in anestesia generale, sotto guida transesofagea (ETE) ed angiografica.
L’esame transesofageo ha confermato la morfologia bilobata a coda di balena con un diametro della landing zone di 19×15 mm e una profondità massima di circa 12 mm, in assenza di trombi.
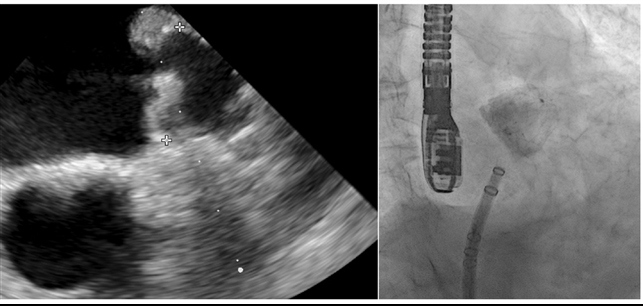
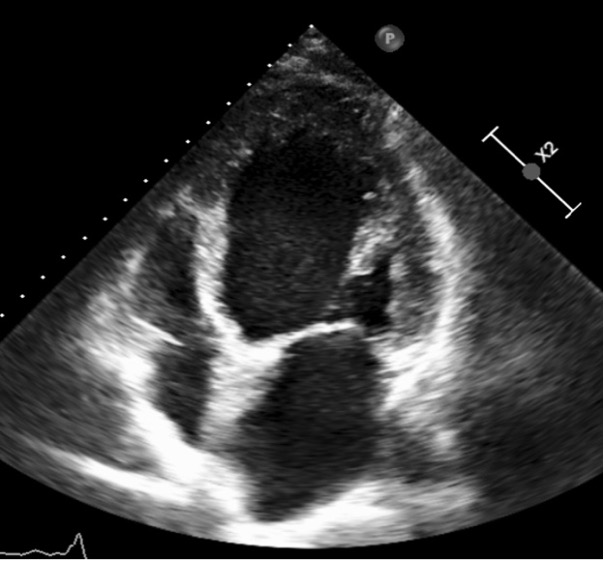
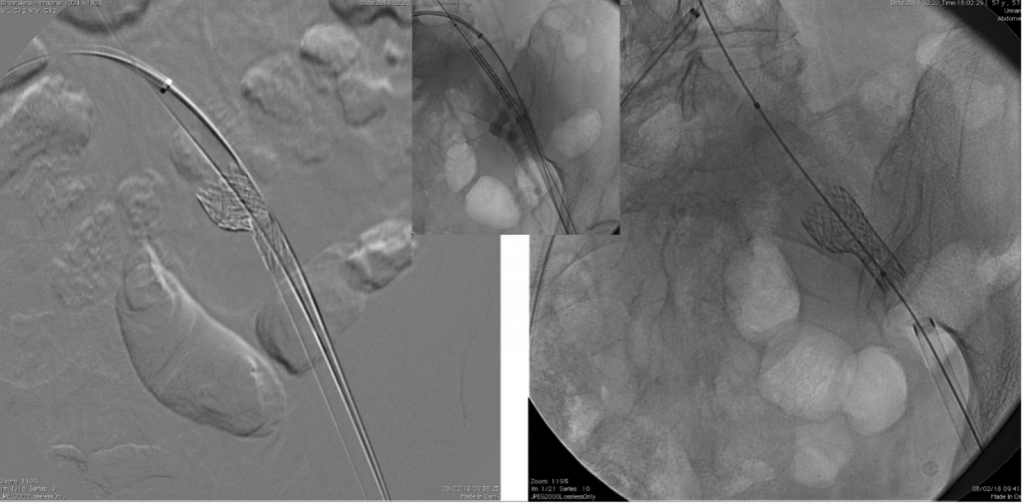
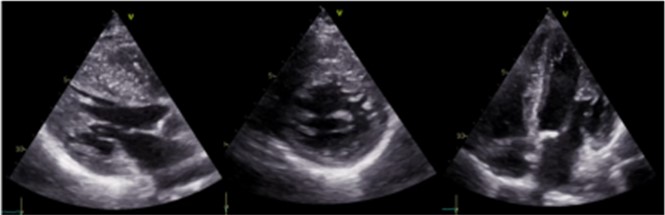
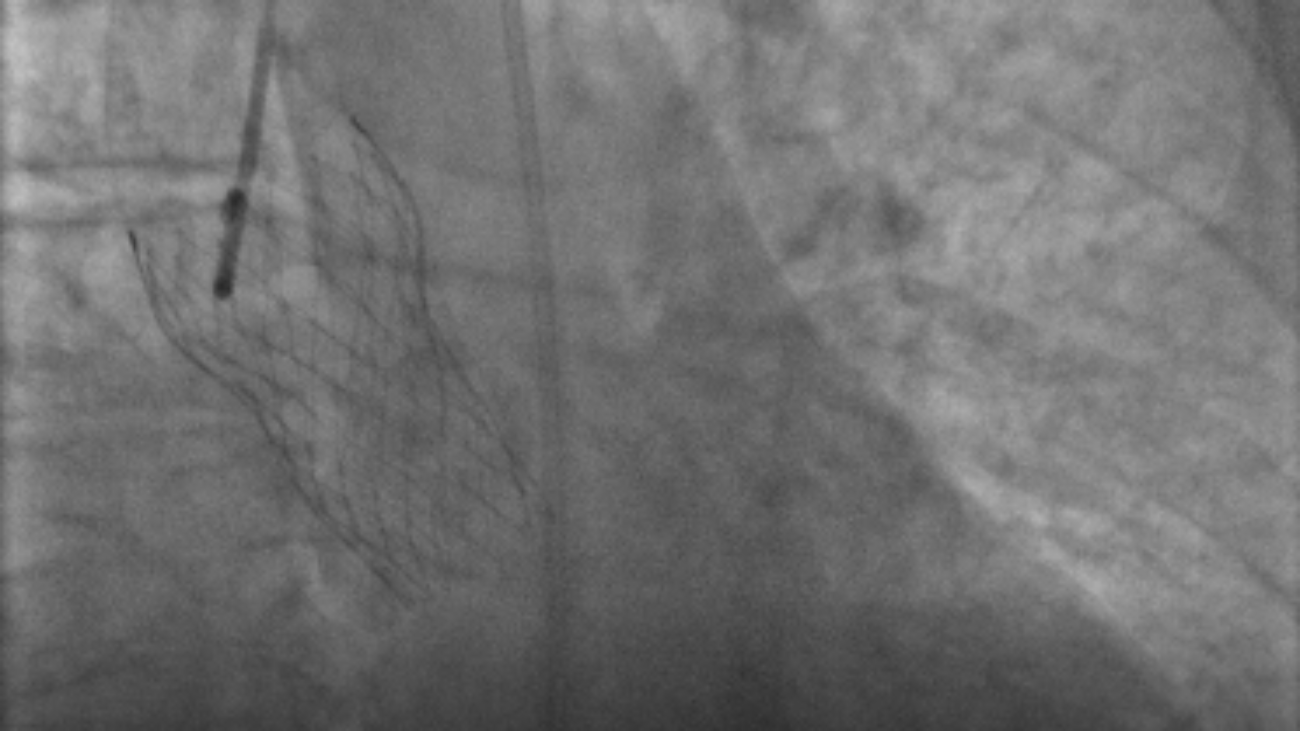
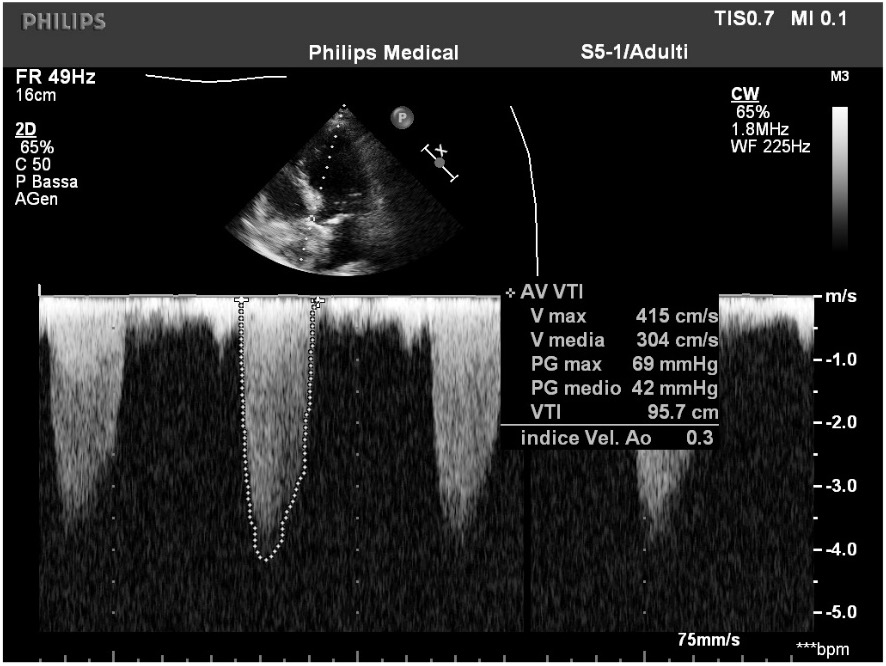
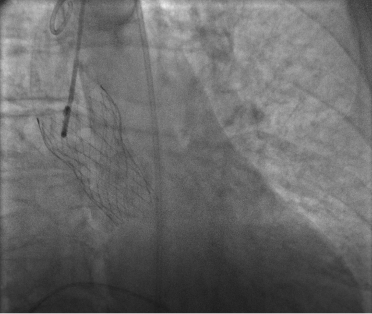
Utilizzando un delivery system di 14F, abbiamo impiantato un Watchman Flex device 24 mm (Boston Scientific, Marlborough, MA, USA) nella porzione prossimale dell’appendice atriale sinistra, come pianificato dalla piattaforma. Dopo due tentativi di impianto prossimale, il device risultava posizionato verso il lobo inferiore, lasciando un ampio leak intorno al lobo opposto. Spingendolo verso l’ostio dell’auricola, con una delicata trazione si è ottenuta una posizione centrale, simmetrica e stabile al “push and pull” test. La posizione finale del device e la deformazione sono state confermate sia tramite ecografia transesofagea che all’angiografia, con un risultato simile a quello predetto da FEops (Figura 2). Il decorso clinico post-procedurale si è svolto in modo regolare, il paziente è stato dimesso a domicilio due giorni dopo la procedura. In considerazione della storia di elevata diatesi emorragica veniva impostata terapia domiciliare con Cardioaspirina a seguito di tre mesi di doppia terapia antiaggregante. Al controllo eseguito tramite ETE a a 45 giorni, il device risultava nella corretta posizione, senza evidenza di leaks né embolizzazione o trombi (Figura 3)
Discussione
Nella pratica comune, la selezione dei device per chiusura di auricola sinistra è ancora basata su misurazioni ecocardiografiche 2D, sebbene l’uso della TC stia assumendo sempre di più un ruolo cruciale nel planning pre-procedurale data la sua non invasività e l’entità di informazioni che fornisce4,5. Per ottenere un’analisi ancora più completa, FEops Heart guide ha sviluppato una piattaforma di simulazione online che permette di riprodurre l’interazione tra diversi device e la specifica anatomia del paziente, applicabile a diversi setting quali ad esempio la sostituzione percutanea di valvola aortica o la chiusura di appendice atriale sinistra6.
Il nostro caso mostra i vantaggi di utilizzare questa piattaforma di simulazione in una delle morfologie più complesse di auricola sinistra, la cosiddetta auricola a coda di balena, con una profondità di impianto di soli 12 mm. Un device con un lobo e un disco, quale l’Amplatzer Amulet è stato già adoperato con successo in presenza di tale anatomia e descritto in letteratura7. Tuttavia, il nostro scenario non offriva profondità sufficiente per questa “sandwich technique”, con il rischio di successivo prolasso del dispositivo. In considerazione dell’analisi eseguita dal software veniva consigliato l’utilizzo di un dispositivo ball-shape quale il Watchman FLX. Tale device infatti durante l’impianto tende inizialmente a lasciare un leak e, una volta rilasciato, deve essere posizionato adeguatamente nella porzione centrale dell’ostio dell’auricola rischiando sotto-compressione e instabilità. FEops ha rappresentato un punto di forza del planning pre- procedurale del nostro paziente, in quanto ha permesso non solo di selezionare il tipo e la misura del device ma anche il giusto posizionamento per evitare leakage.

Le auricole con anatomie sfavorevoli si associano ad un elevato rischio di procedure lunghe, complesse ed economicamente dispendiose, spesso con la necessità di testare molteplici device al fine di scegliere quello più adeguato. L’esecuzione di una corretta pianificazione avvalendosi di tutte le metodiche a disposizione dovrebbe essere indipendente dall’esperienza dell’operatore. Come dimostrato dal nostro centro, un aiuto può derivare anche dai modellini 3D ottenuti dalle scansioni TC volume rendered che permettono impianti personalizzati8.
Quello descritto è il primo caso di auricola con anatomia a coda di balena trattato mediante un ball shape device. Anche se al momento non ci sono trials sull’applicazione di FEops HEARTguide alla selezione dei Watchman Flex devices, questo caso mostra nuovi spunti sulla possibile applicazione di tale piattaforma agli scenari più complessi ai fini di evitare mismatches, errori procedurali e ottenere i migliori outcome clinici.
References
1. Gerhard Hindricks, Tatjana Potpara, Nikolaos Dagres, Elena Arbelo, Jeroen J Bax, Carina Blomström-Lundqvist, et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur Heart J. 2021;42(5):373-498. doi:10.1093/eurheartj/ehaa612
2. Beigel R, Wunderlich NC, Ho SY, Arsanjani R, Siegel RJ. The Left Atrial Appendage: Anatomy, Function, and Noninvasive Evaluation. JACC Cardiovasc Imaging. 2014;7(12):1251-1265. doi:10.1016/j.jcmg.2014.08.009
3. Garot P, Iriart X, Aminian A, Kefer J, Freixa X, Cruz-Gonzalez I, et al. Value of FEops HEARTguide patient-specific computational simulations in the planning of left atrial appendage closure with the Amplatzer Amulet closure device: rationale and design of the PREDICT-LAA study. Open Heart. 2020;7(2):e001326. doi:10.1136/openhrt-2020-001326
5. Kaafarani M, Saw J, Daniels M, Song T, Rollet M, Kesinovic S, et al. Role of CT imaging in left atrial appendage occlusion for the WATCHMANTM device. Cardiovasc Diagn Ther Vol 10 No 1 Febr 2020 Cardiovasc Diagn Ther Percutaneous Struct Valvular Heart Dis Interv. Published online 2020. Accessed January 1, 2020. https://cdt.amegroups.com/article/view/36144
6. Bavo AM, Wilkins BT, Garot P, De Bock S, Saw J, Søndergaard L, et al. Validation of a computational model aiming to optimize preprocedural planning in percutaneous left atrial appendage closure. J Cardiovasc Comput Tomogr. 2020;14(2):149-154. doi:10.1016/j.jcct.2019.08.010
7. Freixa X, Panaro A, Carballo J. Percutaneous Closure of a “Whale Tail” Left Atrial Appendage. Rev Esp Cardiol Engl Ed. 2017;70(9):770. doi:10.1016/j.rec.2016.12.004
8. Robinson SS, Alaie S, Sidoti H, Auge J, Baskaran L, Avilés-Fernández K, et al. Patient-specific design of a soft occluder for the left atrial appendage. Nat Biomed Eng. 2018;2(1):8-16. doi:10.1038/s41551-017-0180-z